Si vous arrivez
directement sur cette page, sachez que ce travail est
un rapport d'étudiants et doit être pris comme tel. Il
peut donc comporter des imperfections ou des
imprécisions que le lecteur doit admettre et donc
supporter. Il a été réalisé pendant la période de
formation et constitue avant-tout un travail de
compilation bibliographique, d'initiation et d'analyse
sur des thématiques associées aux technologies
biomédicales. Nous ne
faisons aucun usage commercial et la duplication
est libre. Si vous avez des raisons de contester
ce droit d'usage, merci de
nous en faire part . L'objectif de la
présentation sur le Web est de permettre l'accès à
l'information et d'augmenter ainsi les échanges
professionnels. En cas d'usage du document, n'oubliez
pas de le citer comme source bibliographique. Bonne
lecture...
Travail d’analyse d’activités de maintenance liées
aux mésusages des DM en exploitation
Les hôpitaux tels que le
CHU de Rouen sont confrontés à la mauvaise utilisation
des dispositifs médicaux. Ces mésusages se rencontrent
principalement en phase d’utilisation du cycle de vie du
dispositif. Dès lors, il devient difficile pour le
service biomédical de pouvoir assurer une gestion des
risques efficace sans le soutien de partenaires
indispensables comme la direction des soins et la
direction de la qualité. Néanmoins cette synergie ne
doit pas masquer le fait que des mesures préventives
doivent être prises à chaque étape de la vie du
dispositif médical, de sa conception à sa destruction.
Mots clés : dispositif médical, mésusage, cycle
de vie , gestion des risques
ABSTRACT
Hospitals such as the
Rouen CHU are often faced with the misuse of the medical
devices wich occurs mainly during use of the life cycle
phase of the device. From that moment on, it becomes
hard for the biomedical service to ensure effective
management of the risks without the essential support of
the medical department of care and quality.However, this
essential synergy must not cover the fact that
preventive measures must have to be taken at every steps
of the life of the device, from its conception to its
destruction.
Key words : device, misuse, life cycle,
management of the risks
Un projet ne se construit pas seul. Il est le fruit d’échanges,
d’aides, de soutiens et de conseils.
Je tiens donc à remercier sincèrement toutes les personnes qui m’ont
permis de mener à bien ce projet :
M. Gilbert FARGES, Docteur-Ingénieur, enseignant chercheur et
conseiller scientifique de la formation ABIH (Assistant Biomédical en
Ingénierie Hospitalière) à l'UTC de Compiègne.
M. Pol-Manoel FELAN, responsable pédagogique de la formation
ABIH à l'UTC de Compiègne.
M.
Alain Donadey, chef de projets et responsable de formation à
l'UTC de Compiègne.
Mme Nathalie MOUTONNET, assistante administrative de la
formation ABIH à l'UTC de Compiègne.
Je tiens à remercier Monsieur Grosjean, Ingénieur
gestionnaire du service biomédical pour son accueil et son aide à
l’intégration dans l’équipe.
Je souhaite également adresser mes remerciements aux ingénieurs et
techniciens du service biomédical de Rouen ainsi qu’aux secrétaires
et logisticien pour leur accueil chaleureux.
L’ensemble du personnel du CHU de Rouen pour leur disponibilité et
professionnalisme
Les différents prestataires du service biomédical du CHU de Rouen
pour leur retour d’expérience et leur disponibilité
Un remerciement particulier pour
Julien et Jean Batiste, techniciens biomédicaux, qui m’ont
fait partager leurs expériences sur les respirateurs
Didier,Stéphane et Adrien pour leur retour
d'expérience sur la dialyse,l'échographie et les contrôles
qualité en radiologie.
L'ensemble du groupe ABIH 2019 pour l'entente
conviviale et ma famille pour sa patiente
Mes collègues du biomédical de Pontoise pour
avoir palié à mon absence pendant cette formation
Je dédie ce travail à toutes ces personnes qui ont œuvré à
l’aboutissement de ce projet.
Le cycle de vie d’un dispositif médical passe par
différentes phases évolutives qui débutent par la conception dans un
bureau d’étude jusqu’à la destruction dans une filière de recyclage.
Tout au long de son parcours, le dispositif doit répondre à un
fonctionnement optimal selon des caractéristiques définies par le
constructeur en fonction de normes scrupuleuses et d’exigences
essentielles pour l’obtention du marquage CE.
Néanmoins, il arrive que des dispositifs médicaux engendrent des
problèmes de mauvaises utilisations qui peuvent parfois avoir de
graves conséquences pour le patient. Ces mésusages peuvent être
notamment le fruit d’une mauvaise conception ou d'une mauvaise
interprétation visuelle du dispositif médical comme de pratiques
professionnelles parfois non respectées ou incorrectement
appliquées.
De ce fait, ils doivent être traités différemment selon qu’ils
relèvent d’une matériovigilance quand le risque clinique pour le
patient est avéré ou bien par des rappels de bonnes pratiques
lorsque les recommandations du constructeur, du service biomédical
ou encore de l’équipe opérationnelle d’hygiène hospitalière ne sont
pas suivies.
Ces mésusages génèrent des problèmes sécuritaires pour le patient
mais également organisationnels et financiers pour l’ensemble des
acteurs et notamment le service biomédical, gestionnaire et
prestataire de service du parc des dispositifs médicaux.
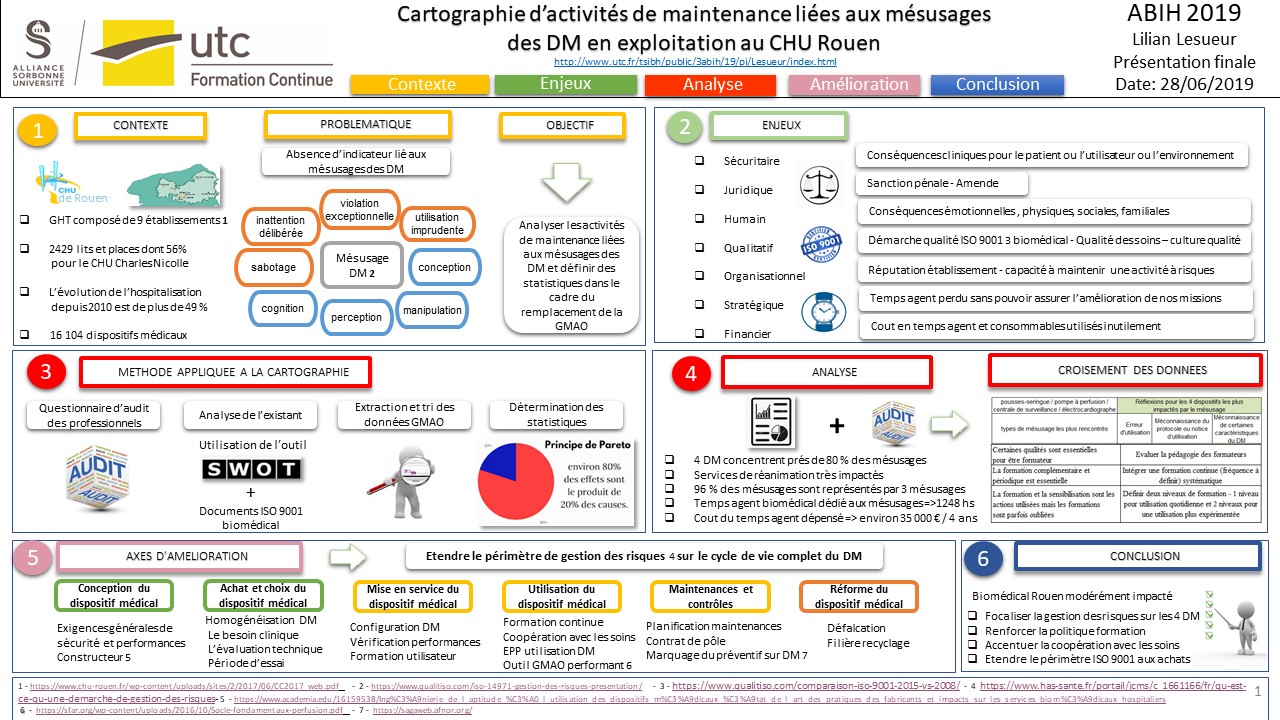
Ce projet a pour objectif de cartographier l’ensemble des mésusages
liés à la maintenance des dispositifs médicaux en phase
d’exploitation. Le recensement de ces informations est assuré par
les techniciens biomédicaux au travers de la GMAO actuelle du CHU de Rouen qui
sera, par ailleurs, prochainement remplacée par un outil plus
performant.
Ce travail d’analyse permettra de bénéficier d’indicateurs de
référence de la situation existante et de pouvoir définir et mesurer
notamment les différents types de mésusages sur les dispositifs
médicaux.
Nous tenterons d’en tirer des enseignements pertinents afin de
définir ou redéfinir des stratégies à adopter dans le cadre de la
gestion des risques, à chaque étape de la vie du dispositif médical.
Les CHU ont été créés par ordonnance du 30 décembre 1958.
Leur conception remonte à 1955, quand sous l'autorité du Pr Robert
Debré, les deux institutions, l'hôpital et de la faculté, sont
devenues partenaires pour donner naissance au CHU par convention
constitutive.
A Rouen, cette convention a été signée le 21 juillet 1965, pour le
centre hospitalier régional, par le Président du conseil
d'administration, M. Bernard Tissot, maire de Rouen et pour l'école
nationale de médecine et pharmacie, par son directeur, le Pr Jean
Fleury, représentant le recteur. On peut donc fixer à l'été 1965 la
naissance officielle du centre hospitalier et universitaire de
Rouen. Dès l'année suivante, l'école nationale est devenue une
faculté et une université a été créée à Rouen associant lettres,
sciences, médecine et droit.
On lui donna le nom de Charles Nicolle en hommage à ce grand
chercheur rouennais, prix Nobel en 1928 pour ses recherches sur le
typhus.[1]
De cette période, il reste une partie de ces bâtiments. Sur les 30
hectares restants plusieurs bâtiments ont été construits au fil des
ans : d'abord l'anneau central et le pavillon Derocque, puis de
nouvelles unités avec par exemple le pavillon mère et enfant dans
les années 1970 ou encore les urgences pédiatriques en 2005. Les
services de plus en plus performants et les découvertes
scientifiques ont donné une réputation internationale à cet
établissement.
Dans les années 1970, les services de soins s'étoffent avec la
création et l'installation du Samu.
Au fil des ans le CHU s'est adapté aux nouvelles technologiques. Le
premier scanner est installé à la fin de cette décennie.
La médecine a fait de grandes avancées ces 20 dernières années avec
les recherches du professeur Éric Mallet sur la mort subite du
nourrisson (1991) ou avec l'invention de la valve aortique du
docteur Cribier (2002).
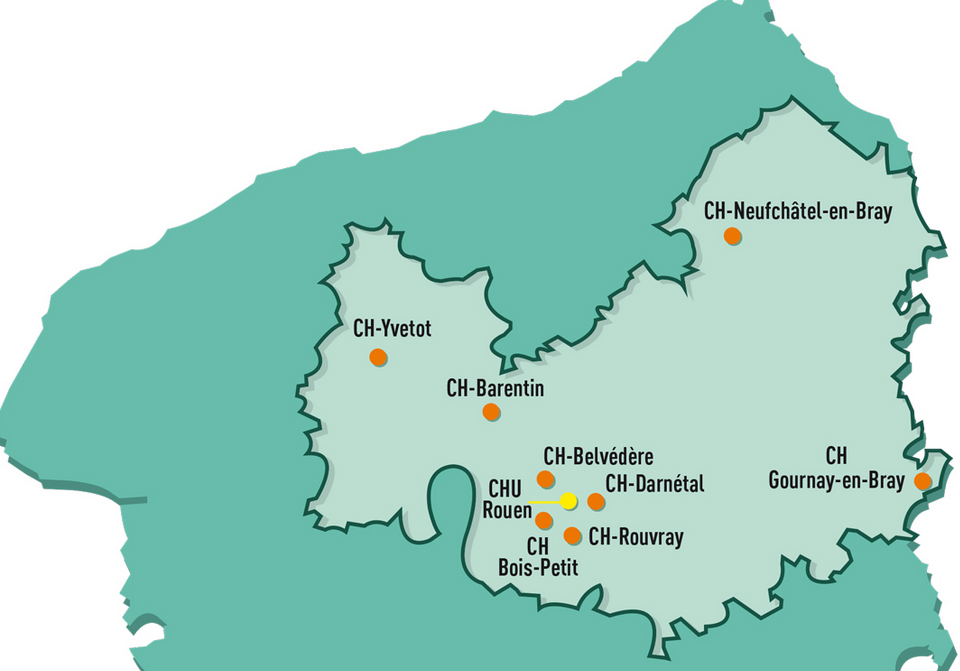
La Directrice générale de l’Agence régionale de santé (ARS) de Normandie
a arrêté vendredi 1er juillet 2016 le périmètre de 11 groupements
hospitaliers de territoire (GHT), composés des 56 établissements
publics de santé et de 11 établissements et services médico-sociaux
publics de la région.
Sa mission est d’améliorer l’accès aux soins hospitaliers pour
les patients sur l’ensemble du territoire, en fonction de leurs
besoins et en développant les complémentarités entre les
établissements sanitaires et médico-sociaux.
Le centre hospitalier universitaire de Rouen est un ensemble de cinq
hôpitaux : l'hôpital Charles Nicolle (Rouen), l'hôpital de
Bois-Guillaume, l'hôpital Saint-Julien (Le Petit-Quevilly),
l'hôpital d'Oissel et l'EHPAD Boucicaut (Mont-Saint-Aignan).
Le CHU de Rouen est le premier employeur de Haute-Normandie avec
plus de 10 000 personnes dont 960 médecins et 600 internes. Le CHU
de Rouen dispose de 2 459 lits répartis sur 5 sites.
Pôle régional de formation des professionnels de santé, le CHU
dispose d'un espace régional des formations des professions de santé
qui regroupe ses 11 écoles paramédicales et accueille 1 500
étudiants. Cet espace de formation assure le lien direct avec la
faculté de médecine et de pharmacie.[2]
L’activité médecine, chirurgie et obstétrique (MCO) représente
506 000 consultations et plus de 160 000 hospitalisations
Le nombre de naissance est de 2777 et le nombre de fécondation in
vitro est de 484.
Le nombre de greffe est de 395 (cardiaque, rénale, moelle
pédiatrique, cornée)
La durée moyenne de séjour (DMS) est de 3.3 jours et environ 261 700
patients sont venus au moins une fois au CHU. Plus 80% des patient
viennent du territoire de santé Rouen/Elbeuf
159 000 passages aux urgences (adultes, pédiatriques et
obstétriques)
Plus de 341 000 appels du SAMU
53 300 séjours en ambulatoire
L’évolution de l’hospitalisation depuis 2010 est de plus de 49 % La répartition des lits
L’ensemble du CHU de Rouen dispose de 2429 lits et places dont 56%
pour le CHU Charles Nicolle
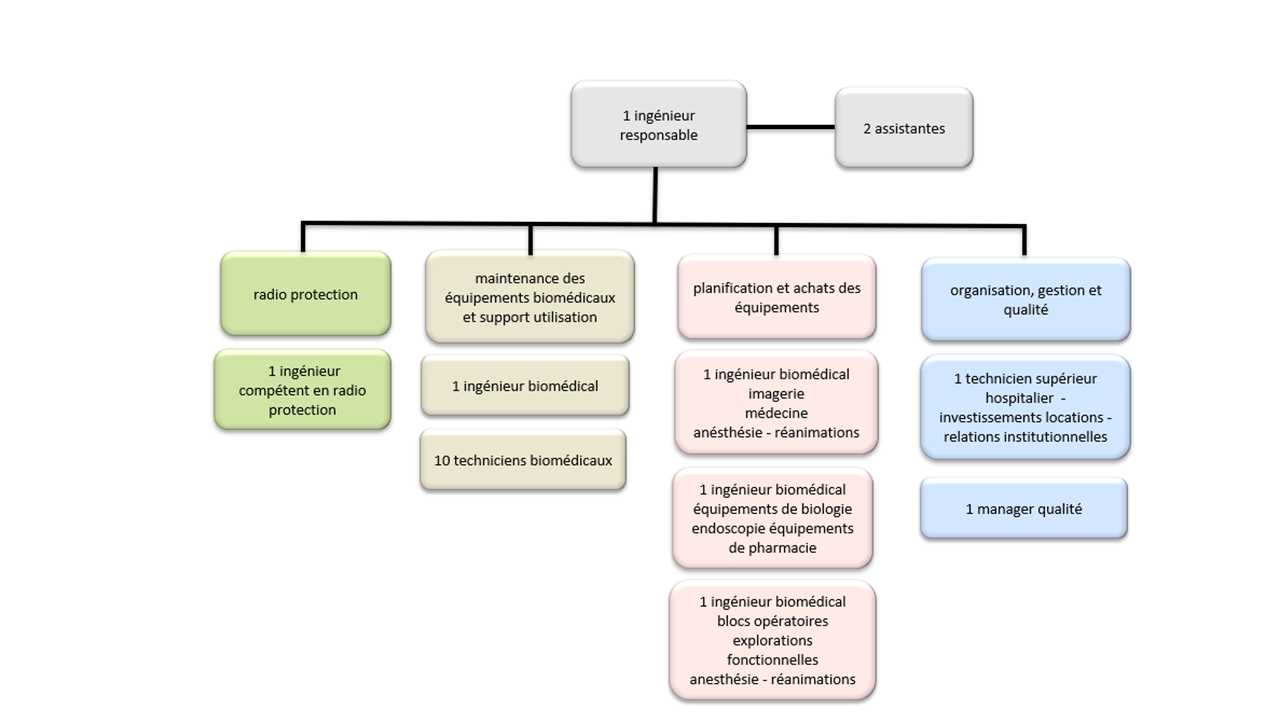
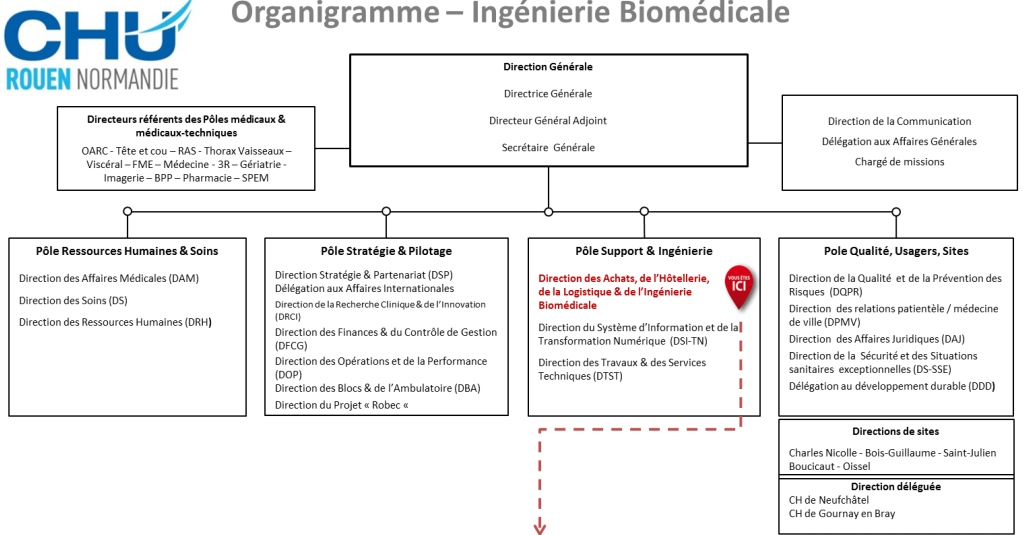
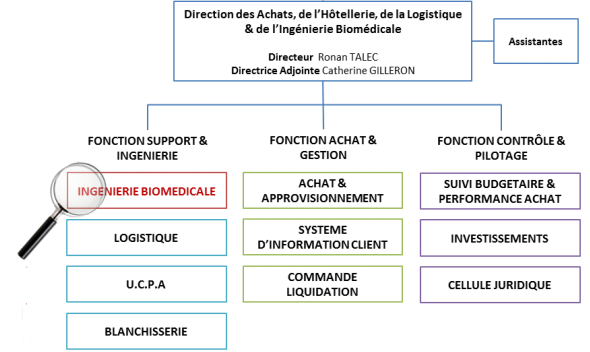
Le service biomédical est rattaché à la direction des achats, de
l’hôtellerie, de la logistique et de l’ingénierie biomédicale (DAHLIB) sous
la direction de M. Ronan Tallec et de son adjointe Mme Catherine
Guilleron.
Le management et la gestion de service
Le service biomédical du CHU de Rouen est géré par M. André
Grosjean, ingénieur, responsable du service. M. Grosjean assure
également la gestion de l’activité de l’atelier « maintenance ».
Les missions et activités du service biomédical
Le service de l’ingénierie biomédicale assure les missions de
conseil, d’expertise, d’achat et de maintenance dans le domaine
biomédical.
Il élaboration et planifie le plan d’équipement médical annuel, en
lien avec :
Il étudie les besoins des services et assure la planification des
achats, il réceptionne le matériel et assure la mise en service du
matériel neuf
Il effectue également la gestion du patrimoine biomédical :
Tenue de l’inventaire et organisation et mise en œuvre de la
maintenance des matériels (curative et préventive)
Il assure la formation des utilisateurs :
Dans le cadre de l’utilisation du matériels neufs
Complémentaire aux matériels en exploitation
Il assure le suivi de la matériovigilance auprès du correspondant
local.
La fonction achat est assurée par 4 ingénieurs adjoints qui couvrent
pour chacun d’eux un périmètre d’activité respectif.
Activité de radioprotection assurée par un conseillé en radio
protection (gestion des contrats de prestations externes dans
son domaine respectif)
Activité d’imagerie, médecine et anesthésie réanimation
Activité de biologie, endoscopie, équipement de pharmacie
Activité de bloc opératoire, explorations fonctionnelles,
anesthésie réanimation
Chaque ingénieur effectue un plan d'investissement annuel qui
représente un total d'environ 1000 équipements à acquérir,
renouveler et installer dans les différentes unités. Chaque
ingénieur est en lien direct avec le pôle concerné afin de
conseiller et d’optimiser le parc de dispositifs médicaux.
Le panel de dispositifs à remplacer ou acquérir est très large
et peu atteindre des montants conséquents. Les achats sont
essentiellement assurés par certains opérateurs nationaux tels que l’UGAP, UNIHA ou le RESAH et
occasionnellement en fonction des procédures du code des marchés
publics (AO, MAPA…).
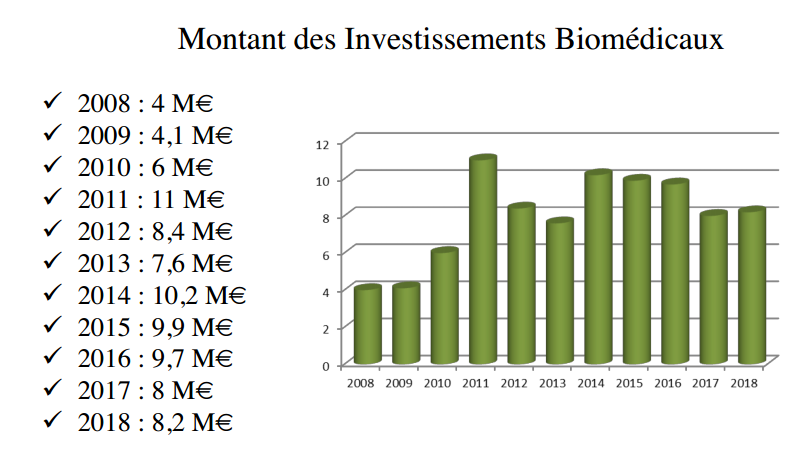
Le patrimoine immobilier biomédical du CHU de Rouen est évalué à 109
M€.
10 techniciens biomédicaux assurent les activités de
maintenances correctives et préventives sur l’ensemble du GHT. Les
techniciens biomédicaux sont chargés d’assurer, sous l’autorité de
l’ingénieur responsable de la maintenance, la maintenance préventive
et curative du parc biomédical réparties sur les 5 sites du CHU de
Rouen, A cela, il convient d’ajouter différentes annexes telles que
l’USMP (prison
Bonne Nouvelle), le centre de Lutte Anti Tuberculose (LAT) et le centre
de rétention administrative.
Le service biomédical est composé de techniciens spécialisés et
organisés en binômes. En 2018, ce sont plus de 8 000 interventions,
curatives et préventives qui ont été réalisées. La part de la
maintenance préventive s’élevait alors à 39%, taux en évolution
constante depuis plusieurs années.
La première certification du service biomédical du CHU de Rouen
selon la norme ISO 9002 a été obtenue en décembre 1999 dans sa
version initiale de 1994. Le dernier audit de renouvellement obtenue
en février 2018 a permis au service biomédical d’obtenir la
certification selon la version ISO 9001-2015.
L’organisme certificateur intervient un fois / an pour un audit de
suivi du système qualité. En interne, des audits sont effectués
chaque année soit par des auditeurs du service biomédical soit par
des auditeurs certifiés de l’établissement.
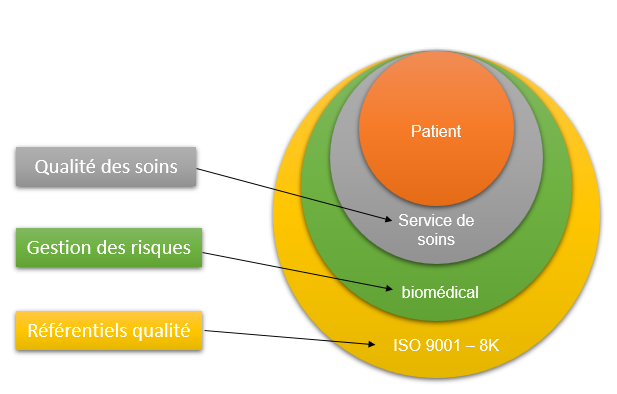
Le service biomédical du CHU de Rouen est certifié ISO 9001 sur le
périmètre de la maintenance et de la formation aux utilisateurs.
Cette norme repose sur un certain nombre de principes de management
de la qualité, notamment une forte orientation client, la motivation
et l’engagement de la direction, l’approche processus et
l’amélioration continue.
La norme ISO 9001-2015 permet de s’assurer que les clients (services
utilisateurs) obtiennent des produits et services conformes et de
bonne qualité.
Les principaux atouts de la nouvelle version ISO 9001-2015 :
Plus d’importance dans l’implication de la direction
Une approche par les risques qui aide à traiter les risques et
opportunités
Structure et termes communs aux autres normes de système de
management
Moins d’exigences pour les procédures documentées
Évaluation globale du contexte du service qui permet de mieux
établir les parties intéressées du service et des
attentes
Prise en compte de toutes les parties intéressées pertinentes
Quel que soit le périmètre de certification des services
biomédicaux, cette norme permet d’adopter des principes de
management et de tirer profit des avantages de la norme de façon à
être plus efficace et ainsi mettre en place des méthodes de travail
plus efficientes.
Le contrat de prestations biomédicales
Dans le cadre de la certification ISO 9001-2015, les prestations de
service font l'objet d'engagements entre la direction des
équipements biomédicaux et les services de pole.
Un contrat est donc établi entre le service biomédical et chaque
pole respectif. Un bon contrat doit satisfaire les deux parties. Ce
contrat doit permettre de déterminer, pour les deux parties, quels
sont les engagements à minima à respecter afin que le système
qualité de la direction des équipements biomédicaux soit maîtrisé.
Les engagements du service biomédical
Prise en charge des interventions et des situations urgentes
Informations sur la demande – possibilité de suivre l’état de
l’intervention via une application informatisée
Réception et reprise du dispositif médical - Tous les jours
ouvrables de 8h30 à 10h30 et de 13h00 à 15h30
Formation complémentaire du personnel utilisateur - A la
demande du service du pôle, une formation complémentaire sur un
dispositif médical déjà en fonction peut est assurée.
Prêt d'un DM - A la demande du service du pôle, un DM peut
être prêté dans la limite des disponibilités
Engagement des services du pôle
Demande d'intervention par l'intermédiaire de l'application
informatique dédiée – Remplir les informations nécessaires au
traitement de la demande
Préparation du dispositif médical avant intervention - mise à
disposition du technicien biomédical ou du fournisseur du
dispositif médical concerné. Le dispositif médical doit être
propre afin de ne pas exposer les techniciens et les
intervenants extérieurs à des risques infectieux.
Déclaration de désinfection pour les matériels issus des blocs
opératoires
Reprise des DM au biomédical - le service récupère ses DM
déposés au biomédical dans les meilleurs délais après clôture de
l'intervention par le biomédical
Suivi d'intervention d'un fournisseur sur un dispositif
médical sous garantie ou sous contrat - Le service du pôle
informe de l'intervention d'un fournisseur lors d'une
intervention sous garantie ou sous contrat -Il fait parvenir le
rapport d'intervention à l'ingénieur biomédical responsable de
la maintenance.
Retour d'un DM de prêt – retour indispensable des DM de prêt
dans un maximum de 2 mois effectif
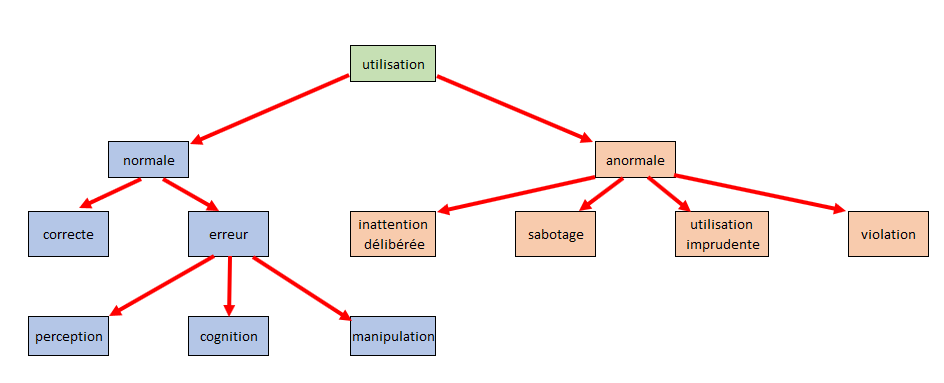
L’utilisation anormale ne peut être sécurisée par le fabricant,
c’est notamment le cas lors :
D’une violation exceptionnelle (utilisation complètement
inappropriée du dispositif)
D’une inattention délibérée aux contre-indications
(l’utilisateur ne tient pas compte des informations relatives à
la sécurité)
D’une utilisation imprudente (sans tenir compte des phénomènes
dangereux)
D’un sabotage ou dégradation volontaire
L’utilisation normale se fait conformément à l’emploi prévu, tant
sur l’utilisation du DM que sur les conditions (formation effectuée,
instructions d’utilisation prises en comptes,). Mais même dans ce
cas une utilisation incorrecte peut survenir notamment en cas
d’erreur :
De perception (ex : alarme non remarquée)
De cognition (ex : valeur numérique mal interprétée)
Ce deuxième type de mésusage est à déclarer dans le cade de la
matériovigilance dans le cas où un risque clinique potentiel pour le
patient peut apparaitre mais aussi un risque pour les soignants,
l’entourage, l’environnement.
La matériovigilance
C’est la surveillance du fonctionnement des dispositifs médicaux et
de leur usage correct par les utilisateurs, en dehors de ceux
utilisés dans des essais cliniques.
La matériovigilance a pour objectif d’éviter que ne se produisent ou
se reproduisent des incidents et risques d’incidents mettant en
cause des dispositifs médicaux, en prenant les mesures préventives
et/ou correctives appropriées. [4]
Les utilisateurs de dispositifs médicaux (usagers, professionnels de
santé, etc.) ont l'obligation de déclarer les incidents qu’ils
rencontrent à la suite de l’utilisation d’un dispositif médical, y
compris lorsque ces incidents résultent d’un mésusage,
c’est-à-dire d’un usage non conforme à la destination ou aux
prescriptions d’utilisation du DM. [5]
Un incident grave se définit comme tout dysfonctionnement ou toute
altération des caractéristiques et/ou des performances d’un
dispositif ainsi que toute inadéquation de l’étiquetage ou de la
notice d’instructions susceptibles d’entraîner ou d’avoir entraîné
la mort ou une dégradation grave de l’état de santé d’un patient ou
d’un utilisateur
Le règlement 2017/745 modifie les règles de matériovigilance
La
base de données Eudamed permet d’améliorer la lisibilité des
dispositifs mis sur le marché. Elle contient notamment les données
réglementaires accessibles aux autorités compétentes afin de leur
permettre d’accomplir les tâches de surveillance du marché qui leur
incombent au titre de la directive n°93/42/CEE
Les fabricants devront notifier à l’ANSM via la base de données Eudamed les
éléments suivants :
Tout incident grave (sauf ceux concernés par les rapports de
tendance)
Toute mesure corrective de sécurité prise à l’égard d’un
dispositif présent sur le marché de l’union européenne
La notification sera faite dès l’établissement de l’imputabilité et
en tout état de cause dans les délais.
La base de données européenne Eudamed, qui doit être effective en
mars 2020, devrait permettre d'assurer un meilleur accès à
l'information sur l'enregistrement, la vigilance et la surveillance
des dispositifs.[6]
Selon le code L 5211-1 de la santé publique : « On entend par
dispositif médical "tout instrument, appareil, équipement,
matière, produit, à l'exception des produits d'origine humaine, ou
autre article utilisé seul ou en association, y compris les
accessoires et logiciels nécessaires au bon fonctionnement de
celui-ci, destiné par le fabricant à être utilisé chez l'homme à
des fins médicales et dont l'action principale voulue n'est pas
obtenue par des moyens pharmacologiques ou immunologiques ni par
métabolisme, mais dont la fonction peut être assistée par de tels
moyens".
Le logiciel constitue également un dispositif médical destiné à être
utilisé spécifiquement à des fins diagnostiques ou thérapeutiques.
Les dispositifs médicaux qui sont conçus pour être implantés en
totalité ou en partie dans le corps humain ou placés dans un orifice
naturel, et qui dépendent pour leur bon fonctionnement d'une source
d'énergie électrique ou de toute source d'énergie autre que celle
qui est générée directement par le corps humain ou la pesanteur,
sont dénommés dispositifs médicaux implantables actifs. »
Cette définition bien que généraliste encadre explicitement la
finalité des dispositifs médicaux : diagnostiquer, prévenir, traiter
une maladie, une blessure ou un handicap, étudier ou remplacer ou
modifier l'anatomie ou un processus physiologique.
Afin de garantir l’ensemble de ces objectifs, il est essentiel que
les processus de conception de dispositif médicaux soient
parfaitement maitrisés et que la connaissance nécessaire à
l’utilisation soit pleinement transmise. C’est le rôle du fabricant
de dispositif médical
Pour cela, il peut s’appuyer sur différentes normes telles que :
NF EN ISO 14 971
La normes ISO 14 971 est une norme harmonisée aux exigences de
la directive européenne sur les dispositifs médicaux. Elle décrit un
processus permettant aux fabricants d'identifier les phénomènes
dangereux associés à son dispositif médical et à ses accessoires,
d'estimer et d'évaluer les risques associés à ces phénomènes
dangereux, de maîtriser ces risques et de surveiller l'efficacité de
cette maîtrise.
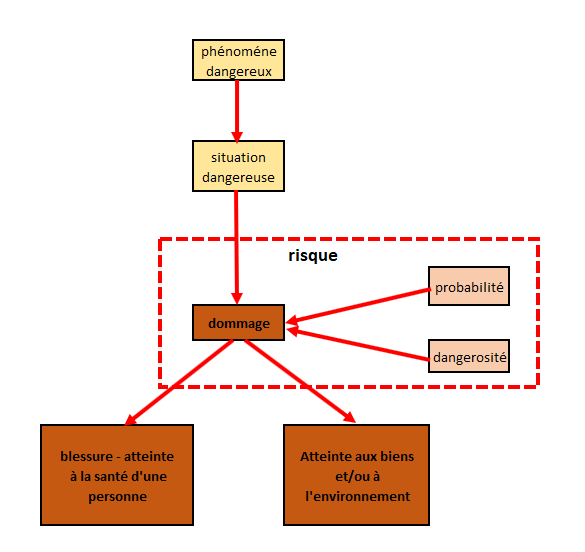
L’ISO 14 971 introduit plusieurs notions permettant de comprendre la
notion de risque :
Le risque est lié à un phénomène dangereux, souvent une
défaillance du dispositif ou un mésusage
Sous certaines conditions peut naître une situation dangereuse
: des personnes, des biens ou l’environnement sont alors exposés
au phénomène dangereux. Cette situation dangereuse peut se
traduire par des dommages : blessure ou atteinte à la santé, à
l’environnement ou aux biens
Ces dommages sont caractérisés par leur probabilité
d’occurrence et leur gravité : c’est cette combinaison qui
définit la notion de risque.
Utilité de la norme en vue de l’obtention du marquage CE d’un
DM
Les grands principes de l’ISO 14971 pour la gestion des
risques
Place de la gestion des risques dans le cycle de vie du
dispositif médical
L’ISO 14971 n’est pas obligatoire mais elle est reconnue comme moyen
de réponse à divers exigences Européenne. Elle est systématiquement
mise en œuvre par les fabricants de dispositifs médicaux car mettre
en place une activité de gestion des risques est impérative pour
pouvoir marquer « CE
» un produit.
La directive 93/42/CEE encadre les modalités de certification CE des
dispositifs médicaux. Elle définit les différentes procédures de
marquage CE, applicables en fonction de la classe des dispositifs
médicaux.
Le point commun de toutes ces procédures est la gestion d’une
documentation technique incluant les résultats de l’activité de
gestion des risques. [7]
Évolution de la réglementation
Le règlement 2017/745 remplacera la directive 93/42/CEE en 2024. Il
prévoit l’utilisation de seuils et indicateurs dans le cadre de la
surveillance après commercialisation (SAC)
Le plan de surveillance après commercialisation comprend au moins
des indicateurs et des seuils adaptés pour procéder à la
réévaluation continue de l’analyse bénéfice/risque et de la gestion
des risques afin de mettre à jour le processus de gestion des
risques.
Dépassement de seuil et analyse de tendance
Analyse périodique et évènement inattendu
Moyenne mobile et répartition des données
Les données sont comparées à une valeur moyenne mobile, c’est à
dire calculée sur la base des dernières données.
Les outils utilisés sont :
Le tableau : utile pour afficher plusieurs dimensions mais
assez peu lisible
Les secteurs : pour distinguer le poids de chaque catégorie
mais finalement assez peu lisible surtout lorsque les catégories
sont nombreuses
L’histogramme : certainement la plus fine des représentations
(exemple : diagramme de Pareto)
Les fabricants seront donc soumis à de nouvelles contraintes de
gestion des risques sur leur produit mais uniquement dans leur
périmètre d’intervention.
Afin de tendre vers une parfaite maitrise de gestion des risques et
des mésusages, l’exploitant pourra s’appuyer sur différentes normes
et guides :
La norme ISO 9001 : 2015 - levier central pour le management de
la gestion des risques
La nouvelle certification ISO 9001 dont bénéficie le service
biomédical de Rouen oriente le système de management de la qualité
vers l'efficience.
La gestion des risques est une approche préventive. Elle est un
élément principal de la nouvelle version de l’ISO 9001. La bonne
gestion de la démarche qualité devient le résultat de la bonne
gestion des risques.
La gestion des risques, induit la prise en compte de la capacité à
fournir un produit ou service conforme dans la durée. La mise en
application de la gestion des risques à l’ensemble du système de
management de la qualité (SMQ), aux processus et aux activités est donc
devenue essentielle.
Dans la mise en œuvre de la gestion des risques, les principales
étapes sont :
L’identification des risques,
Leur quantification,
L’identification des risques “majeurs” (principaux,
prépondérants ou majoritaires)
La gestion des risques est aussi une manière d’œuvrer pour
l’amélioration continue : les actions préventives répondent à un
risque. Il s’agit d’une non-conformité potentielle mais non encore
survenue.
En opposition, une action corrective est mise en place en face à un
risque mal identifié ou mal géré qui a débouché sur un
dysfonctionnement, un produit non-conforme …
Les risques ne peuvent pas être dissociées des opportunités. En
effet, la norme ISO 9001-2015 développe le concept d’incertitude
positive. Elle permet d’intégrer dans les SMQ les éléments plus
positifs et tout en améliorant le système.
Les opportunités peuvent être définies comme « Toute occasion
favorable qui peut aboutir à l’amélioration des résultats ou des
performances du système ».
On n'écrit pas une procédure pour qu'elle soit lue ou appliquée, pas
plus qu'on ne l'écrit pour que chacun fasse la même chose. On écrit
une procédure parce qu'on a identifié que c'était la réponse la
mieux adaptée à la maîtrise d'un risque donné car il y a souvent
plusieurs moyens de maîtriser le risque en question.
Il faut systématiquement se poser la question des risques qui pèsent
sur le système, sur ce qui peut l'empêcher d'atteindre ses
objectifs.[8]. La gestion des risques est une
exigence additionnelle qui ne se substitue en rien aux concepts de
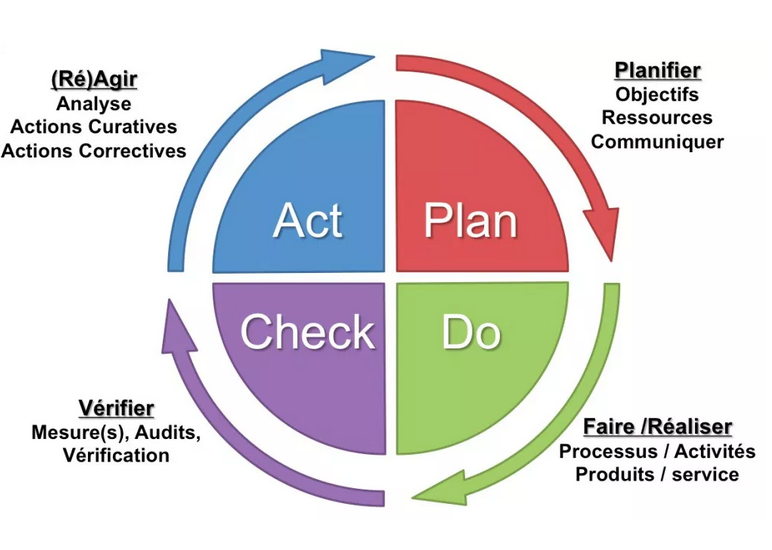
la qualité : l’approche processus et la roue de Deming ou PDCA.
Il s’agit de reproduire continuellement 4 étapes pour mener des
actions. Elle est un des piliers des démarches Qualité. C’est un des
éléments de base de l’amélioration continue. [9]
Les systèmes de management normalisés existants dans le domaine de
la qualité comme l’ISO 9001 sont des outils méthodologiques éprouvés
pour maîtriser les risques propres à ces domaines mais le liant
nécessaire au management global des risques n’est pas abordé. D’où
l’intérêt de se tourner vers la norme ISO 31000.
L’ISO 31000 complète les référentiels existants dans le cadre d’une
démarche de management intégré et rationnel. Cette norme n’est
d’ailleurs pas « certifiable ». Il s’agit d’un outil complémentaire
permettant de développer le principe de l’approche risque et de
l’intégrer dans les pratiques managériales.
La mise en œuvre de l’ISO 31000 permet notamment :
D’augmenter la probabilité que les objectifs seront atteints
et d’encourager un management proactif et prendre conscience de
la nécessité d’identifier et de traiter le risque.
D’améliorer l’identification des opportunités et des menaces
et de se conformer aux obligations légales, réglementaires,
ainsi qu’aux normes internationales
D’améliorer la rédaction des rapports obligatoires et
volontaires et d’améliorer la gouvernance, d’accroître
l’assurance et la confiance des parties prenantes
D’établir une base fiable pour la prise de décision et la
planification et d’améliorer les moyens de maitrise
D’améliorer l’efficacité et l’efficience opérationnelle,
De renforcer les performances en matière de santé et de
sécurité, d’améliorer la prévention des pertes et le management
des incidents, de minimiser les pertes.
Autant de facteurs qui permettent un management proactif face aux
risques encourus. L’ISO 31000 est à considérer comme un outil qui,
en complément des référentiels de management, permet de cadrer
l’approche risque, d’optimiser notamment le pilotage des processus
et l’atteinte des objectifs [10]
La norme NF S99-172
La norme NF S99-172 est intéressante pour les services biomédicaux
car elle permet de développer un système de management du risque lié
à l’exploitation des DM. Elle assure la fusion de normes
internationales de management de la qualité telle que l’ISO
9001-2015 ou la norme ISO 31000 sur le management du risque.
Les exigences de la norme NF S99-172 sont adaptées de celles des
normes ISO 9001 et ISO 31000 avec toutefois la création de deux
éléments notables :
Une entité compétente chargée du management du risque
comprenant au moins un représentant de la direction avec pouvoir
de décision afin de garantir la capacité d’action rapide et
pertinente
Un Dossier de Maîtrise Continue des Risques (DMCR) »
alimenté par un système de gestion des informations documentées
apportant les preuves sur les résultats de son efficacité et
performance.
Le guide des bonnes pratiques – Maintenance des DM – obligations
et recommandations
Ce guide « métier » rédigé par un groupe de professionnel composé
d’ingénieurs biomédicaux, de correspondants matériovigilances, de
juristes et de directeurs apporte un focus notamment sur la gestion
des risques lors de :
L’acquisition d’un DM
La mise en service du DM
Ainsi que le développement d’une des méthodes d’analyse des risques
existants : la méthode AMDEC
Les différentes étapes de la gestion des risques
Identification des risques
L’identification consiste à recenser toutes les parties exposées au
risque. Le service biomédical établie une liste contenant tous les
risques potentiels liés aux DM. Il doit distinguer les risques les
plus importants d’un côté et les moins importants d’un autre côté et
analyser leur corrélation.
Évaluation des risques
Il faut évaluer et hiérarchiser les risques en fonction de leur
gravité, déterminer leurs impacts potentiels et l’étendue des
préjudices. Déterminer les causes.
Définition des solutions
Le service biomédical dispose des solutions envisageables et doit
utiliser la plus adaptée. Il peut définir la solution en fonction du
risque lui-même en étudiant la possibilité d’une élimination ou
d’une limitation de ses effets.
Mise en œuvre des solutions
Après avoir déterminé la solution la plus adaptée, il faut
déterminer des actions préventives et évaluer les moyens mis en
application.
Le contrôle
La gestion des risques nécessite un suivi régulier. Ce suivi vise à
garantir la fiabilité de chaque étape. Cela permet de mettre en
place des solutions préventives à moyen et à long terme. Une
évaluation des résultats est nécessaire. [11]
Certifié ISO 9001 depuis de nombreuses années, le service biomédical
a su développer des stratégies afin de définir des moyens de
maitriser les problèmes d’utilisation.
Le passage vers la nouvelle version ISO 9001 en 2018 axée davantage
sur le traitement de la gestion des risques nécessite de faire un
état des lieux sur le mésusage afin de savoir si les actions mises
en place sont suffisantes et efficaces
Le service biomédical du CHU de Rouen souhaite donc effectuer une
cartographie de l’ensemble des mésusages recensés au quotidien par
les techniciens dans les interventions de maintenance de la GMAO.
Ce travail d’analyse pourra permettre d’enrichir la prochaine GMAO «
Carl® » qui doit venir remplacer l’actuel logiciel devenu trop
limité dans ses fonctionnalités et sa praticité.
Nous allons donc effectuer dans un premier temps une analyse
matricielle avec l’outil SWOT afin de définir les forces et faiblesses
liées à la mise en place du projet de cartographie des mésusages.
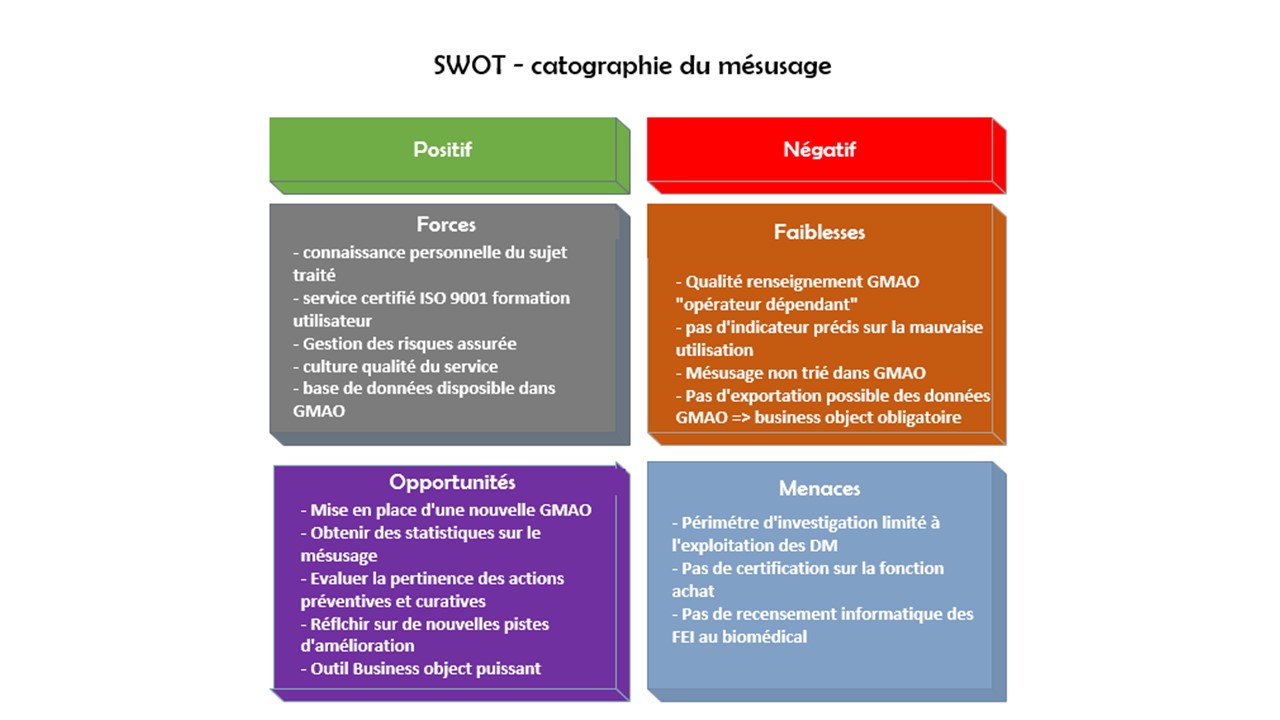
Bonne connaissance du sujet traité : Il n’existe pas un
service biomédical qui ne soit pas confronté un jour ou l’autre
par le mésusage des DM – Je suis particulièrement sensible à
cette problématique que j’ai déjà eu l’occasion de traiter dans
d’autres domaines professionnels.
Certification ISO 9001 : la certification ISO 9001 repose sur
un certain nombre de principes de management de la qualité,
notamment une forte orientation client, la motivation et
l’engagement de la direction, l’approche processus et
l’amélioration continue. La bonne gestion de la démarche qualité
devient le résultat de la bonne gestion des risques.
Gestion des risques : Le management des risques, autrement dit
l’approche par les risques et opportunités, est une exigence
nouvelle de la norme ISO 9001-2015. Il est essentiel d’aborder
la gestion des risques à travers l’identification préalable des
résultats attendus.
Culture qualité du service : Le servie biomédical est dans sa
huitième certification sur 4 versions différentes (ISO 9002-1994
à ISO 9001-2015). L’ensemble des acteurs du service biomédical
sont directement impliqués dans le management de la qualité et
sont des acteurs à part entière dans leur domaine de compétence
respectifs – chacun gère son processus.
Base de données existante : Une GMAO est utilisée pour le
traitement des interventions curatives et préventives des DM.
Une codification des interventions est effective selon le type
de maintenance effectuée ou les problèmes rencontrés – certaines
interventions sont enregistrées sous des codes spécifiques
lorsqu’une mauvaise utilisation est constatée par le technicien.
Faiblesse
Qualité des renseignements GMAO « opérateur dépendant » : Les
interventions sont renseignées et classées par code – la
précision des renseignements dédiés aux mésusages est
essentielles afin de cibler précisément la problématique et de
pouvoir l’exploiter – la multitude des codes existants peut
induire des erreurs d’aiguillage et certaines interventions
liées aux mésusages peuvent se retrouver noyées dans la masse
des interventions curatives ou préventives.
Absence d’indicateur sur la mauvaise utilisation des DM : Les
informations fournies par codes dans la GMAO peuvent être
utilisables pour définir des indicateurs de suivi et d’alerte
sur les mésusages – ces indicateurs sont néanmoins tributaires
de la précision des renseignements fournis par les techniciens
ou les utilisateurs.
Mésusage non trié : Il n’existe pas, par ailleurs, de
types ou de familles regroupant ou définissant les mésusages ce
qui rend difficile la pertinence d’indicateur et la
hiérarchisation dans l’état actuel de la GMAO.
Pas d’exportation possible des données : il n’existe pas dans
le GMAO actuelle de possibilité d’extraction des données sous
Excel ou autre logiciel. La seule solution est d’utiliser le
logiciel « Business Object » en créant les requêtes nécessaires
à l’exportation des bonnes données. Ce logiciel est néanmoins
très puissant à la condition d’être très précis dans la création
des requêtes.
Mise en place d’une nouvelle GMAO : Une nouvelle GMAO est
toujours un moment important dans l’organisation d’un service
biomédical – Une hiérarchisation préalable des mésusages
pourrait permettre de créer des familles de mésusage. Un champ
dédié aux mésusages pourrait être intégré dans la GMAO avec une
liste déroulante par famille de mésusage.
Obtenir des statistiques : Des indicateurs pertinents d’alerte
et de suivi pourraient ainsi être mis en place grâce à la
hiérarchisation des mésusages dans la nouvelle GMAO
Evaluer la pertinence des actions effectuées : Un comparatif
ou suivi annuel voire pluriannuel de ces statistiques
permettrait d’objectiver la pertinence des actions préventives
ou correctives effectuées contre le problème de mésusage.
Réfléchir à de nouvelles pistes d’amélioration : Dans le cas
ou la récurrence des problèmes de mésusage est mise en évidence
par les indicateurs, il conviendrait de redéfinir ou renforcer
les actions existantes.
Menaces
Périmètre d’analyse limité à l’exploitation : Une maitrise
complète de la gestion des risques liée aux mésusages ne peut
être pleinement efficace que si elle englobe l’ensemble du
processus de la vie d’un dispositif médical – le périmètre à
analyser définie pour le projet se situe en phase d’exploitation
néanmoins nous aborderons les autres phases du DM, de façon plus
théorique, afin de couvrir l’ensemble des risques liés aux
mésusages.
Certification de la fonction achat : Cette fonction importante
dans la lutte contre le mésusage ne fait pas partie du périmètre
de certification ISO 9001 du service biomédical néanmoins des
actions pertinentes sont effectives pour garantir un matériel
conforme aux exigences des utilisateurs et du service
biomédical.
Déclaration interne matériovigilance : L’ingénieur biomédical
en charge du traitement des déclaration de matériovigilance doit
traiter ces documents de façon matérialisé. La gestion de ces
déclarations est ensuite effectuée sous fichier Excel mais il
n’est néanmoins pas possible de trier les matériovigilances
liées aux mésusages car les informations ne sont pas saisies par
type ou famille mais par date.
Recensement des FEI : les déclarations d’événements
indésirables sont gérées par la direction de la
qualité/communication qui retransmet aux services concernés les
déclarations sous forme de lien informatique. Ce lien permet de
répondre directement à la problématique rencontrée. Néanmoins,
le service biomédical n’a pas la possibilité de classer
automatiquement ces déclarations pour une faire une base de
données exploitable notamment pour lutter contre le mésusage.
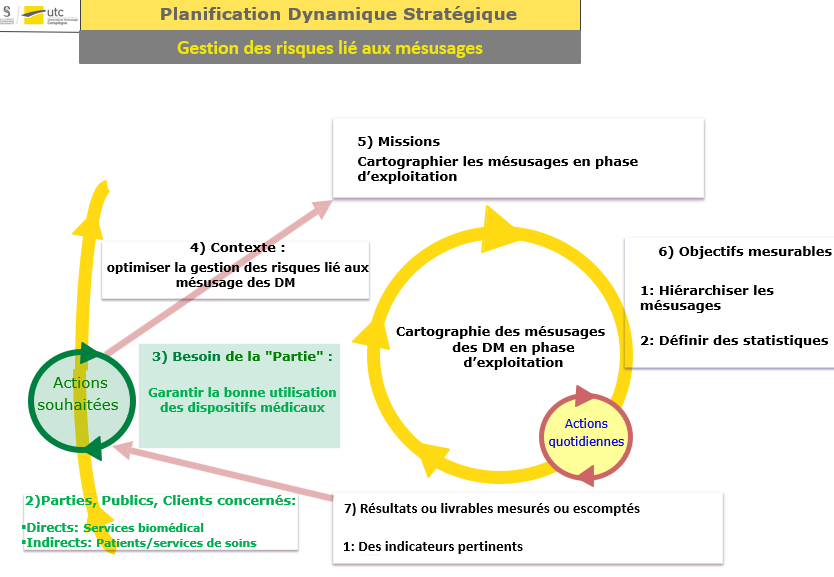
En nous aidant de l’analyse contextuelle effectuée avec la matrice
SWOT, nous allons définir une planification dynamique stratégique
afin de définir clairement notre problématique.
Un dispositif médical est sujet à différents types de mésusage
de sa conception à sa destruction. Il est donc nécessaire de mettre
en place des barrières de sécurité à chaque étape de sa vie afin de
maitriser les risques à priori et à postériori.
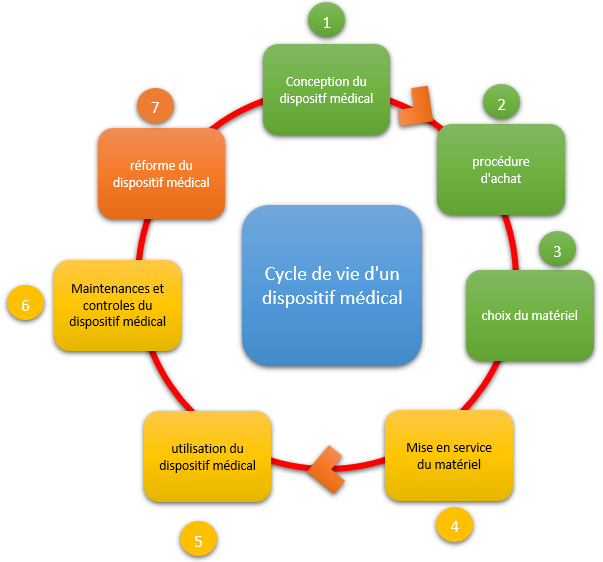
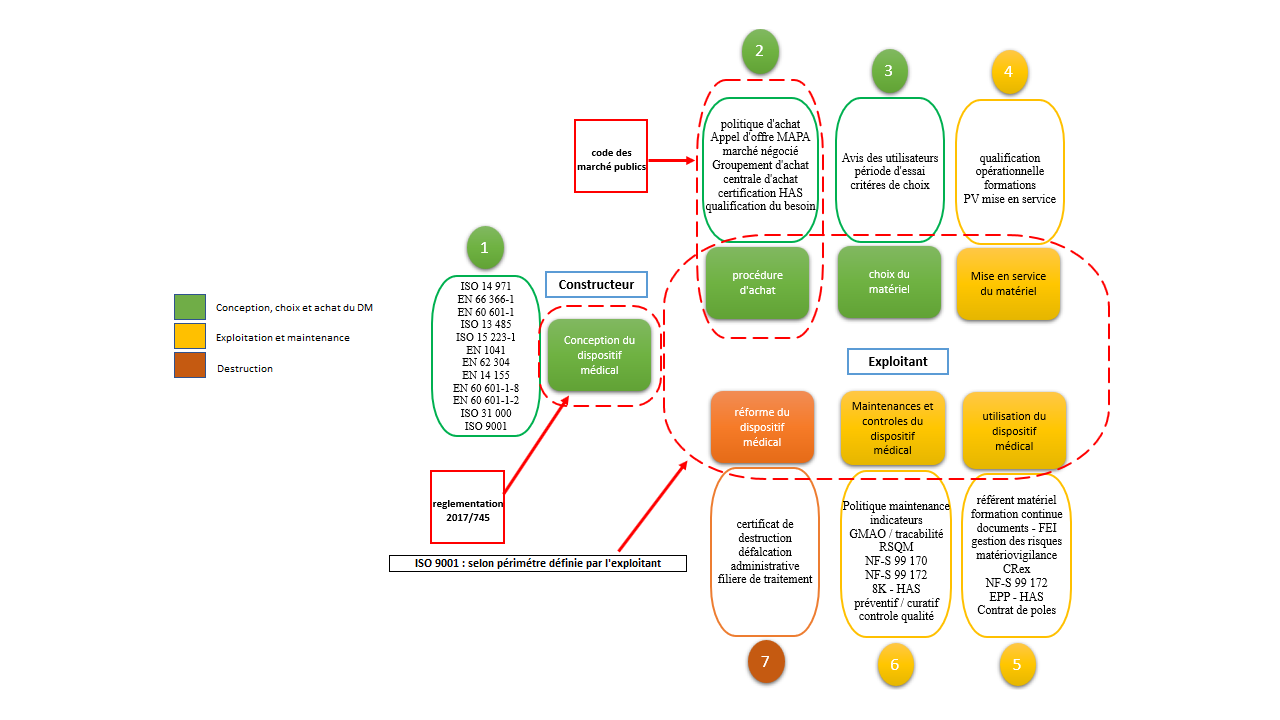
La vie d’un dispositif médical se compose de 3 parties distincts :
Le processus d’acquisition qui englobe 3 phases : la conception, l’achat et le choix du DM
Le processus d’exploitation composé de 3 phases : la mise en service, l’utilisation et la
maintenance
Le processus de destruction par la
mise en réforme administrative du DM
Phase de conception
Dédiée aux constructeurs et bureaux d’étude de DM – cette phase doit
répondre à des cahiers des charges et des normes très précises en
vue du marquage CE pour une commercialisation en Europe. Des normes
applicables aux dispositifs sont essentielles pour minimiser le
risque lié aux mésusages des DM
Phase d’achat
Étape très importante pour le service biomédical qui va définir
précisément son besoin en lien direct avec l’utilisateur. C’est à
cette étape que commence pour le service biomédical sa longue quête
de lutte contre le mésusage. Ces exigences notamment en termes de
normes, sécurité et performances devront être clairement définies et
explicites dans le cahier des charges. Cette étape décidera ou à
défaut limitera les potentielles conséquences sécuritaires et
financières liées aux mésusages. Choix du matériel
Cette étape est très importante car c’est le moment ou l’on doit
s’assurer que le produit proposé correspond parfaitement aux
exigences du cahier des charges – l’utilisateur est un maillon
incontournable à cette étape car c’est lui qui aura en charge
l’utilisation optimale du DM – tous les critères sont à prendre en
compte (sécuritaires, ergonomiques, cognitifs, qualitatifs…et
financiers) – son avis sera essentiel pour le choix à faire – un
dispositif dont le choix est imposé sera mal utilisé ou très vite
dégradé. Phase de mise en service
Il s’agit du moment de la qualification opérationnelle – les
performances du dispositif sont testées en condition d’utilisation
et les utilisateurs ainsi que les techniciens biomédicaux
bénéficient d’une formation à l’utilisation par le technicien
commercial ou l’ingénieur en charge de la mise en service – les
configurations et les seuils d’alarme sont vérifiés systématiquement
– A l’issue de cette qualification, un procès-verbal de mise en
service est rédigé et signé par les deux parties. La date de début
de garantie du DM commence à prendre effet.
Phase d’utilisation du DM
C’est la phase la plus critique dans la vie du DM. L’appareil est
soumis à différentes contraintes et dérives d’utilisation qu’il faut
très vite analyser afin de préserver l’intégrité du dispositif.
L’analyse des pratiques professionnelles et des dérives sont souvent
source de compréhension des mésusages sur les DM.
Phase d’entretien et de vérification
La maintenance est l’ensemble de toutes les actions techniques,
administratives et de management durant le cycle de vie d'un bien,
destinées à le maintenir ou à le rétablir dans un état dans lequel
il peut accomplir la fonction requise. Ces entretiens préventifs ou
curatifs sont assurés selon la politique maintenance du service
biomédical. Ils sont assurés soit par le service biomédical soit par
le constructeur soit par une société de tierce maintenance. Parfois
un partenariat peut être établie entre le constructeur et le service
biomédical afin de définir les niveaux et types de maintenance
assurés par l’un ou l’autre. – un regard rigoureux est cependant à
apporter au travail effectué par les sociétés externes afin que les
dispositifs soient pleinement opérationnels et puissent retrouver
leur configurations et réglages initiaux. Sur cette phase, les
mésusages sont peu fréquents puisqu’ils n’impliquent pas les
utilisateurs au premier abord… sauf quand certains dispositifs
médicaux disparaissent du service pour des raisons de prêt
inter-services par exemple.
Dès lors la recherche du dispositif pour effectuer les maintenances
préventives, par exemple, devient problématique. Ces pratiques
souvent rencontrées ne sont pas des mésusages en tant que tels mais
sont à prendre en compte dans la gestion des risques surtout quand
les configurations et les seuils sont différents selon les
spécialités.
Phase de réforme du matériel
Un matériel destiné à la réforme doit être identifié comme hors
service et sorti du parc de DM. Cette réforme entraîne une mise à
jour de l'inventaire avec précision de la date de sortie du matériel
sur GMAO.
Dans le cas ou un DM pris en charge par un prestataire devient
irréparable, alors un certificat de destruction doit être fourni au
service biomédical pour acter la réforme
Les possibilités de mésusage sont nombreuses tout au long de la vie
du DM. Notre périmètre d’étude des mésusages est axé sur
l’exploitation des DM néanmoins nous aborderons de manière générale
les possibilités d’amélioration face aux mésusages car la lutte
s’effectue en continue, sur chaque phase de la vie du dispositif, de
manière préventive et curative.
Les enjeux sont nombreux et dépassent de loin le périmètre du
service biomédical.
Enjeux sécuritaires
Le patient est au cœur de nos préoccupations. Il n’est pas
concevable qu’une mauvaise utilisation ou qu’une mauvaise
interprétation d’une information d’un DM puisse avoir des
conséquences cliniques pour le patient. Face à ces accidents trop
fréquents, parfois graves et souvent évitables, les enjeux du
renforcement d’une culture de sécurité sont nombreux.
Enjeux juridiques
Une mauvaise utilisation d’un dispositif médical est susceptible de
causer un dommage au patient. Dans ce cas, celui-ci peut demander à
être indemnisé de son préjudice. L’indemnisation des accidents
médicaux en France repose sur un système complexe, qui tente de
trouver un équilibre entre deux impératifs : d’un côté la nécessité
de dédommager les victimes, dans la matière où les dommages sont
particulièrement graves, et d’un autre côté la volonté d’encourager
les avancées de la médecine et de tenir compte de l’aléa inhérent à
l’activité médicale. La victime peut également saisir la justice
pénale. Dans une grande majorité des cas, cette démarche a pour
objectif de sanctionner le ou les professionnels concernés, ainsi
que l’établissement de santé où a eu lieu le préjudice.
Enjeux qualitatifs
L’émergence d’une culture qualité et sécurité des soins est une
priorité d’action car les fragilités de prise de conscience au
niveau des équipes de soins peuvent alimenter les causes majeures de
mésusage associées aux dispositifs médicaux.
Enjeux humains
Les patients et les familles en termes de conséquences
émotionnelles, physiques, sociales, familiales, mais aussi les
professionnels de santé impliqués dans un mésusage de DM.
Enjeux stratégiques
Réputation de l’établissement, capacité à maintenir et développer
une activité à risques dans le cadre de l’organisation de l’offre de
soins.
Le mésusage des DM impacte sur les organisations des services. Ainsi
le service biomédical dépensera inutilement du temps agent à
réceptionner et contrôler le dispositif puis tracer ces actions dans
le registre de sécurité, qualité et maintenance. Le service de soins
dépensera du temps inutilement à effectuer une demande
d’intervention, trouver un dispositif de prêt, déposer et récupérer
le dispositif au service biomédical. Autant de temps perdu à ne pas
pouvoir assurer l’amélioration des missions quotidiennes.
Enjeux financiers
Outre le cout en temps agent, les consommables utilisés à mauvais
escient sont nombreux et amputent le budget fonctionnel des services
biomédicaux et de soins.
Le mésusage des dispositifs médicaux est une problématique qui
touche l’ensemble des établissements de santé à plus ou moins grande
échelle. Le CHU de Rouen est donc confronté au même titre que
d’autres établissements à ces dérives de pratique et/ou
d’utilisation.
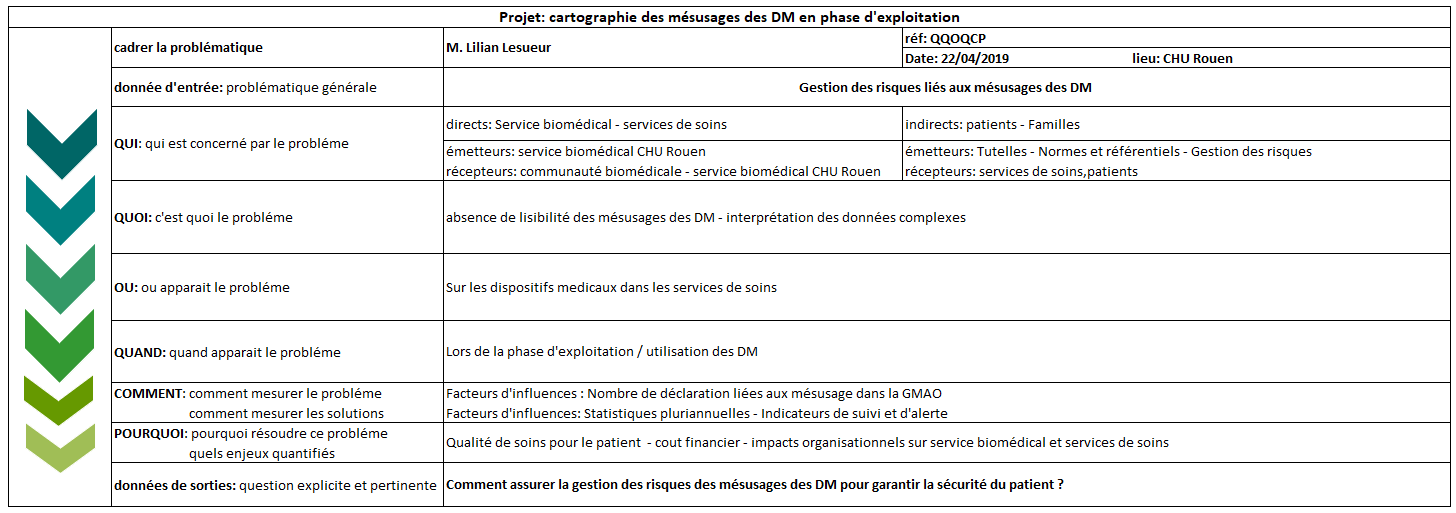
Afin de pouvoir analyser les mésusages du CHU de Rouen, il convient
au préalable de faire un état des lieux de l’existant en matière de
prévention contre le mésusage.
Cet état des lieux nous permettra de définir si les actions mises en
place sont efficaces ou auraient besoin d’être ajustées.
Pour cela, nous effectuerons d'une part un audit auprès des
professionnels de santé et biomédicaux afin de connaitre leur vision
sur le mésusage et les pratiques réalisées. D'autre part, une
analyse de la base de données sera effectuée au travers de la GMAO
existante MP5. Elle permettra de dégager des informations
pertinentes nécessaires à l'étude de notre sujet.
Cette base de données devra ensuite être triée afin d’épurer ce qui
est utile et pertinent de ce qui ne l’est pas.
Il conviendra sans doute de reformuler les commentaires en vue de
pouvoir effectuer par la suite des regroupements par items ou
familles. Enfin des statistiques sous formes de tableaux, graphiques
et indicateurs pourront être élaborés.
Cela nous conduira à pouvoir effectuer des constats en croisant les
réponses de l'audit avec les statistiques déterminées et définir
d'éventuelles pistes d'amélioration exploitables. Nous élargirons
notre périmètre sur l’ensemble de la vie du dispositif médical afin
d’avoir une vue d’ensemble sur ce qu’il est possible d’organiser en
termes de gestion des risques.
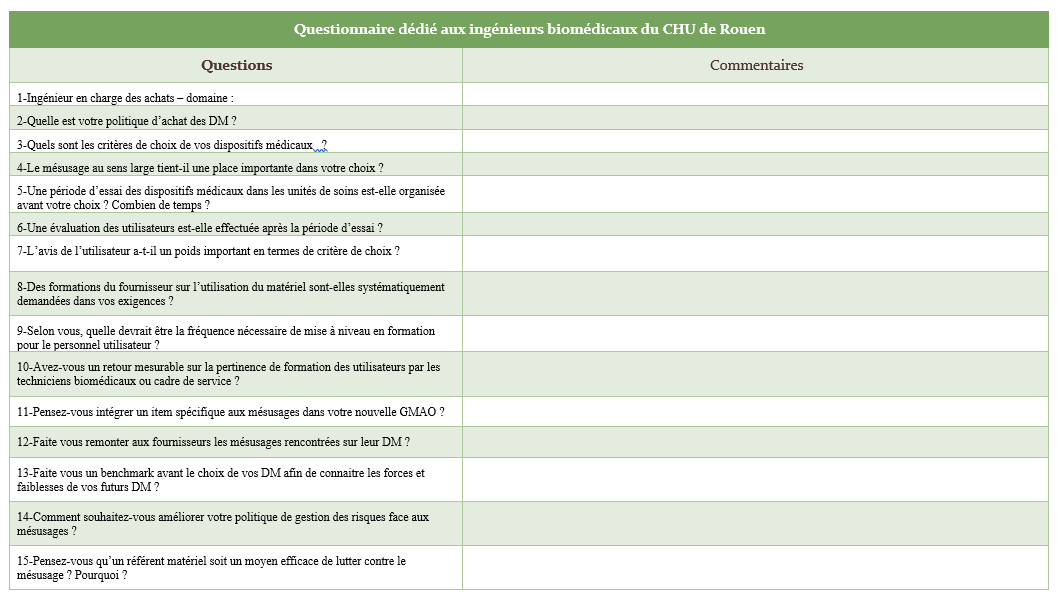
Un questionnaire d’audit a été réalisé et utilisé auprès de
l’ensemble des acteurs en lien avec l’une des étapes de la vie d’un
dispositif médical
2 prestataires externes / fournisseurs pour la partie
conception et exploitation/maintenance
6 techniciens biomédicaux sur 10 pour la partie exploitation
/maintenance
3 Ingénieurs biomédicaux sur 4 pour la partie achat
4 utilisateurs (référents matériel de réanimation) pour la
partie exploitation/utilisation
Ce recueil de renseignements va nous permettre par la suite de
mettre en concordance les statistiques obtenues de l’analyse de la
base de données extrait de la GMAO avec la vision de chacun sur
cette problématique.
Ainsi un questionnaire spécifique non nominatif dédié à chaque corps
de métier regroupe une douzaine de questions. Les résultats sont
représentatifs d’un échantillon de personnel, l’ensemble des
intervenants n’ayant pu être interrogés pour raison de disponibilité
ou de congés.
Il est à noter que l’ensemble des personnes interrogées se sont
prêtées aux jeux des questions/réponses de façon naturelle et
professionnelle.
Les réponses de l’ensemble des personnes auditées ont été
regroupées sous forme de thématiques avec séparation des
commentaires plutôt positifs (en noir) ou plutôt négatifs (en rouge)
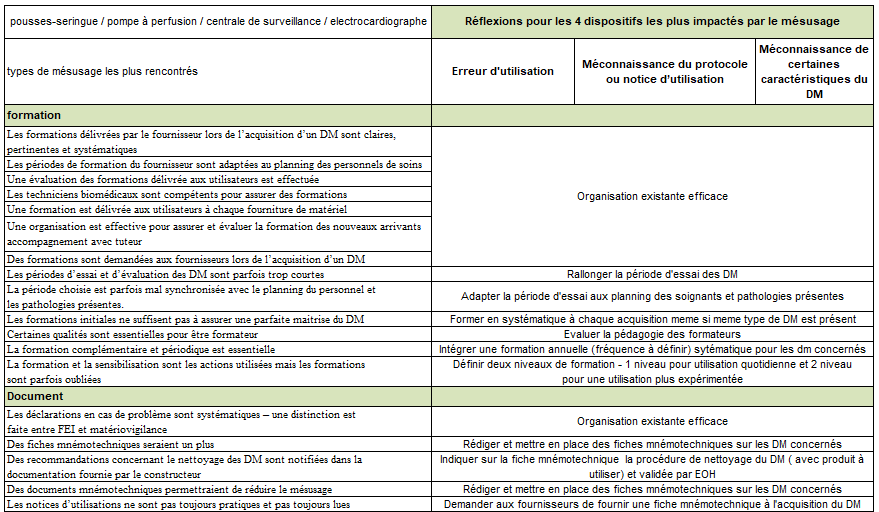
Formation
Les formations délivrées par le fournisseur lors de l’acquisition
d’un DM sont claires, pertinentes et systématiques
Les périodes de formation du fournisseur sont adaptées au planning
des personnels de soins
Une évaluation des formations délivrées aux utilisateurs est
effectuée
Les techniciens biomédicaux sont compétents pour assurer des
formations
Une formation est délivrée aux utilisateurs à chaque fourniture de
matériel
Une organisation est effective pour assurer et évaluer la formation
des nouveaux arrivants – accompagnement avec tuteur
Des formations sont demandées aux fournisseurs lors de l’acquisition
d’un DM
Les périodes d’essais et d’évaluations des DM
sont parfois trop courtes
La période choisie est parfois mal synchronisée avec le planning
du personnel et les pathologies présentes.
Les formations initiales ne suffisent pas à assurer une parfaite
maitrise du DM
Certaines qualités sont essentielles pour être formateur
La formation complémentaire et périodique est essentielle
La formation et la sensibilisation sont les actions utilisées mais
les formations sont parfois oubliées
Documents
Les déclarations en cas de problème sont systématiques – une
distinction est faite entre FEI et matériovigilance
Des fiches mnémotechniques seraient un plus
Des recommandations concernant le nettoyage des DM sont notifiées
dans la documentation fourni par le constructeur
Des documents mnémotechniques permettraient de
réduire le mésusage
Les notices d’utilisations ne sont pas toujours pratiques et pas
toujours lues Caractéristiques des DM
Les DM sont faciles d’utilisation dans la majorité des cas
Certains dispositifs sont plus impactés que d’autres en termes de
mésusage
Des améliorations sont à étudier sur certains
DM en termes d’ergonomie, de poids et d’intuitivité.
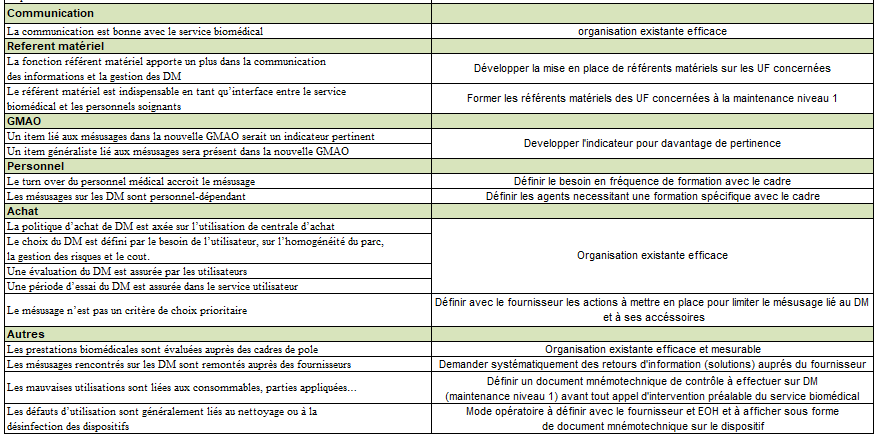
Communication
La communication est bonne avec le service biomédical Référent matériel
La fonction référent matériel apporte un plus dans la communication
des informations et la gestion des DM
Le référent matériel est indispensable en tant qu’interface entre le
service biomédical et les personnels soignants
GMAO
Un item lié aux mésusages dans la nouvelle GMAO serait un indicateur
pertinent
Un item généraliste lié aux mésusages sera
présent dans la nouvelle GMAO
Le personnel
Le turn over du personnel médical accroit le
mésusage
Les mésusages sur les DM sont personnel-dépendant
Achat
La politique d’achat de DM est axée sur l’utilisation de centrale
d’achat
Le choix du DM est défini par le besoin de l’utilisateur, sur
l’homogénéité du parc, la gestion des risques et le cout.
Une évaluation du DM est assurée par les utilisateurs
Une période d’essai du DM est assurée dans le service utilisateur
Le mésusage n’est pas un critère de choix
prioritaire Autres
Les prestations biomédicales sont évaluées auprès des cadres de pole
Les mésusages rencontrés sur les DM sont remontés auprès des
fournisseurs
Les mauvaises utilisations sont liées aux
consommables, parties appliquées, à la mauvaise connaissance du DM
ou de certaines de ses fonctionnalités et également aux
inattentions
Les défauts d’utilisation sont généralement liés au nettoyage ou à
la désinfection des dispositifs
Nous croiserons par la suite les synthèses de l’audit avec
l’état de l’existant et les statistiques issues de l’extraction des
données de la GMAO afin de déterminer les axes d’amélioration
éventuelles.
Afin de recenser de façon assez exhaustive les actions mises en
place par le service biomédical du CHU de Rouen, nous pouvons nous
reposer sur la documentation du management de la qualité ISO
9001-2015 puisque le service est audité chaque année par un
organisme de certification externe.
La phase d’exploitation
La formation des utilisateurs
L’organisation de la prestation biomédicale est fortement axée
sur la satisfaction du client, fondement de l’ISO 9001.
Ainsi, la formation des utilisateurs et la possibilité de demande de
formations complémentaires par les utilisateurs font parties du
périmètre de certification du service biomédical. Par simple demande
via un imprimé dédié, le cadre, s’il le souhaite, peut diligenter
une formation pour son personnel et/ou lui-même. Ces formations
complémentaires peuvent être délivrées par le technicien biomédical
ou par le fournisseur selon les besoins.
Contrat de prestation
Un contrat de prestation est défini entre utilisateurs et
service biomédical. Un bon contrat est un contrat équitable entre
les 2 parties. Des obligations et exigences sont donc
contractualisées et doivent être honorées
Ainsi, les prestations de maintenances sont définies en termes de
délais d’intervention maximum, de traçabilité (exigence
réglementaire) et de possibilité de suivi par le client (GMAO)
notamment.
La GMAO
Des informations sur les dysfonctionnements rencontrés par les
techniciens biomédicaux sur les DM sont incrémentées dans les
commentaires de la GMAO. Ainsi le cadre peut connaitre le
dysfonctionnement inerrant au DM déclaré. Un commentaire écrit,
suite à la demande d’intervention, est toujours rédigé par le
technicien biomédical.
La maintenance préventive et curative
Cette étape importante dans la vie ou plutôt dans le
prolongement de la vie du dispositif médical est l’axe principal
dans le management qualité puisque cette étape est pleinement dans
le périmètre de certification du service biomédical du CHU de Rouen
Les maintenances curatives sont prises en charge dans un délai
contractualisé et les maintenances préventives sont planifiées
annuellement. Chaque technicien a son planning de maintenance
préventive annuelle à disposition.
Compétences et organisation des équipes
La maintenance est assurée par une équipe de 10 techniciens
qualifiés dans un domaine de compétence particulier. Les techniciens
ont un niveau technique BTS (brevet de technicien supérieur) ou
licence. Chaque technicien est accompagné d’un binôme. Les techniciens
composant ce binôme se séparent la gestion des interventions des
dispositifs selon leurs affinités. En cas de congés, le technicien
présent assure l’ensemble de la prise en charge des interventions
dans le domaine de compétence complet du binôme. De ce fait, la
présence d’au moins un des deux techniciens composant le binôme est
requise.
Tout nouvel arrivant est formé par un référent pour lui transmettre
son expérience. Cette formation type « Compagnonnage » est l’un des
éléments majeurs de la formation du nouvel arrivant.
Des formations complémentaires spécifiques sont assurées dans le
cadre de la formation continue ou dans le cadre d’achat de matériel
négocié avec le fournisseur pour maintenir un niveau de compétences
performants et ainsi être en alerte de veille technologique.
Un questionnaire d'évaluation est fourni à chaque participant afin
de vérifier la pertinence de la formation délivrée à l’agent. Une
planification peut être revue si les objectifs n’ont pas été
atteints. Une procédure spécifique encadre ce besoin.
Management de la qualité
Chaque technicien est acteur du management de la qualité du
service biomédical et gère l’intégralité de son processus
Les techniciens biomédicaux participent aux :
réunions du Comité Qualité
réunions plénières
revues de processus et de direction.
Le pilotage et l’animation du comité qualité sont assurés par l’un
des techniciens biomédicaux Suivi des actions qualité
Des Audits internes sont réalisés chaque année afin de mesurer la
qualité de la prestation et la satisfaction du client. La gestion
des non conformités est assurés et des actions correctives sont
délivrées le cas échéant.
Néanmoins le périmètre de certification ne couvre pas l’ensemble des
prestations du service biomédical.
La phase achat et choix du matériel
La fonction achat
Ainsi, la fonction achat n’est pas soumise aux exigences de la
norme car hors périmètre de certification.
Cela n’empêche nullement le service biomédical d’avoir des exigences
qualitatives et sécuritaires en termes d’achats de dispositifs
médicaux.
La politique d’achat du service biomédical est axée selon les
recommandations DGOS qui orientent l’acquisition de
dispositifs médicaux des centres hospitalier plutôt par des
centrales d’achat ou groupement d’achat tels que le RESAH, UNIHA,
UGAP. Cette volonté de maitrise économique n’est pas antagoniste à
une bonne gestion des risques mais elle a néanmoins ces forces et
faiblesses.
Des procédures d’achat sous MAPA ou AO sont également réalisées
selon les montants engagés en cas d’offre incompatible avec le
besoin dans les centrales d’achat.
Le besoin est défini avec l’avis des professionnels et des essais
d’utilisation de DM sont réalisés pendant une période déterminée
dans le service concerné afin de vérifier la concordance de
performance entre le besoin et le produit proposé. Ainsi chaque
dispositif est évalué par l’utilisateur et le choix final s’effectue
en tenant compte des remarques des utilisateurs et des critères de
choix prédéfinis.
Lors de la mise en service du DM, une formation initiale du
prestataire est assurée auprès des utilisateurs et des techniciens
biomédicaux.
Les achats sont assurés par 5 ingénieurs biomédicaux qui ont la
gestion de pôles d’activité spécifiques. Ainsi chaque ingénieur est
référent de pôles en matière d’achat biomédical. La validation des
achats est assurée par l’ingénieur, responsable du service
biomédical. Un ingénieur est dédié, en complément de sa fonction
d’acheteur, à la gestion des déclarations de matériovigilance et des
flux d’information ascendants et descendants entre ANSM et CHU, en
lien avec le référent matériovigilance de l’établissement (le
pharmacien)
La GMAO existante est assez ancienne et ne répond plus aux
besoins de l’évolution de la gestion de maintenance – celle-ci sera
prochainement remplacée par la GMAO Carl.
La première difficulté rencontrée concerne les droits d’accès et la
connaissance du fonctionnement de la GMAO existante. Dans un premier
temps, l’utilisation du logiciel s’est effectuée via le compte d’un
technicien avant que la DSI fournisse un accès temporaire aux données.
Quelques heures de manipulation et d’aide de techniciens permettent
de prendre en main rapidement le fonctionnement et la compréhension
de la GMAO.
Extraction
Les interventions clôturées sont regroupées sous forme de codes qui
correspondent à un type d’intervention. Un tri de ces codes est donc
nécessaire afin d’affiner l’extraction des données utiles.
Il n’est pas possible d’exporter les données de ces codes sous
Excel ou tout autre format de fichier car la fonction « exportation
» n’existe pas.
Après avoir sélectionné les codes et les requêtes pertinentes, le
responsable du service biomédical a pu effectuer une extraction des
données via le logiciel « Business Object » de plus de 4 années
d’archivage afin d’obtenir un fichier exploitable.
Le fichier « brut » ainsi obtenu n’est pas utilisable en tant que
tel et doit être préalablement travaillé afin d’être épuré de
multitude de lignes inutiles telles que :
Doublon ou triplon d’intervention
Code générique
Intervention n’ayant pas de rapport avec le sujet étudié due à
des erreurs d’aiguillage
Intervention sans rapport avec le service biomédical
Pour cela, il faut étudier ligne par ligne les quelques 1600
interventions extraites de la GMAO.
Une fois ce premier travail fastidieux effectué, nous pouvons
rapidement nous apercevoir que la formulation des demandes
d’intervention est parfois peu exploitable par manque de précision.
En effet, il n’est pas rare de rencontrer des demandes du type «
matériel en panne » ou « ne fonctionne pas » ce qui rend difficile
la définition ou désignation du problème à la source.
D’autre part, certaines demandes n’ont rien à voir avec le problème
initial ou le jargon employé n’est pas du tout adapté.
Il devient donc nécessaire de reformuler les demandes quand cela est
possible afin de pouvoir par la suite exploiter les données sous
forme de familles ou d’items. Ce travail redondant s’effectue
également ligne par ligne.
Enfin, les réponses des techniciens sont également à regrouper par
famille afin d’être exploitables. Nous pouvons déterminer un tableau
composé de plusieurs rajouts de colonne.
Les interventions résiduelles peuvent alors être traitées et
sélectionnées selon :
L’année de l’intervention
Le type de dispositif
Le code de l’unité fonctionnelle
Le type de demande (utilisateur)
Le type de problème analysé par le technicien
Le type de mésusage
L’action corrective liée au mésusage (le technicien)
Le nombre d’heure effectuée pour chaque intervention
La valorisation du temps agent
C’est sur la base de ces différentes informations que nous
allons définir des statistiques et indicateurs pertinents afin de
les croiser avec les actions existantes mise en place. Nous
consoliderons ces résultats en les croissant avec les résultats de
l’audit.
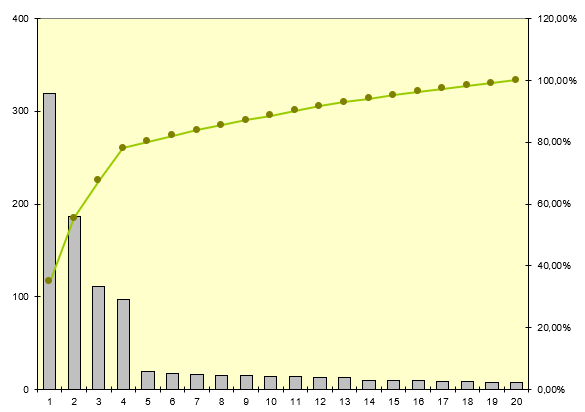
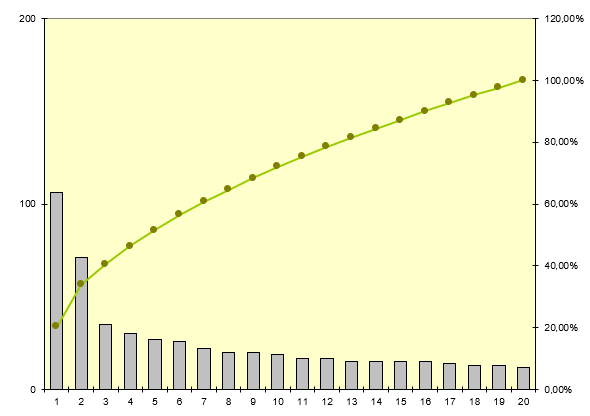
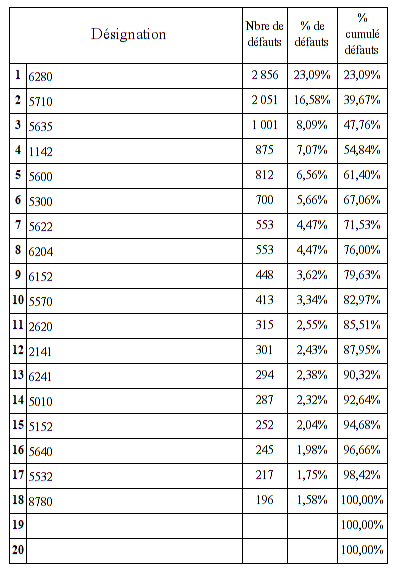
Nous allons pour cela utiliser le diagramme de Pareto - Le
diagramme de Pareto sert à décomposer un problème relativement vaste
en sous-problèmes afin de déterminer lesquels sont prioritaires.
C'est un outil qui permet de visualiser un classement par importance
décroissante de défauts, de causes ou de dysfonctionnements, selon
des critères quantitatifs qui peuvent être la fréquence, le coût, un
nombre d'apparitions de mésusage…Le diagramme de Pareto est
accompagné d'une courbe des valeurs cumulées de toutes les colonnes.
Ce diagramme est construit en plusieurs étapes :
Collecte des données
Classement des données au sein de catégories
Calcul du pourcentage de chaque catégorie par rapport au total
Tri des catégories par ordre d'importance
L’analyse s’effectue sur une période de plus de 4 années soit de
janvier 2015 à mars 2019 sur la base des interventions renseignées
par les techniciens biomédicaux dans la GMAO existante.
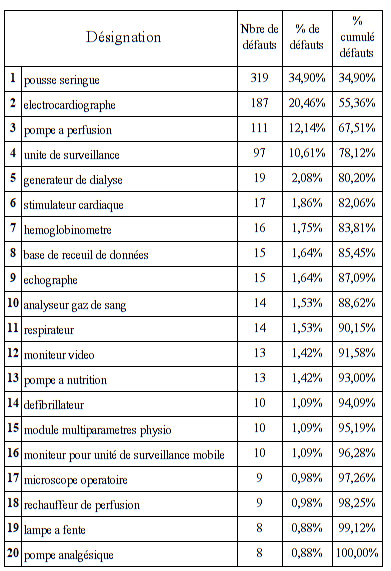
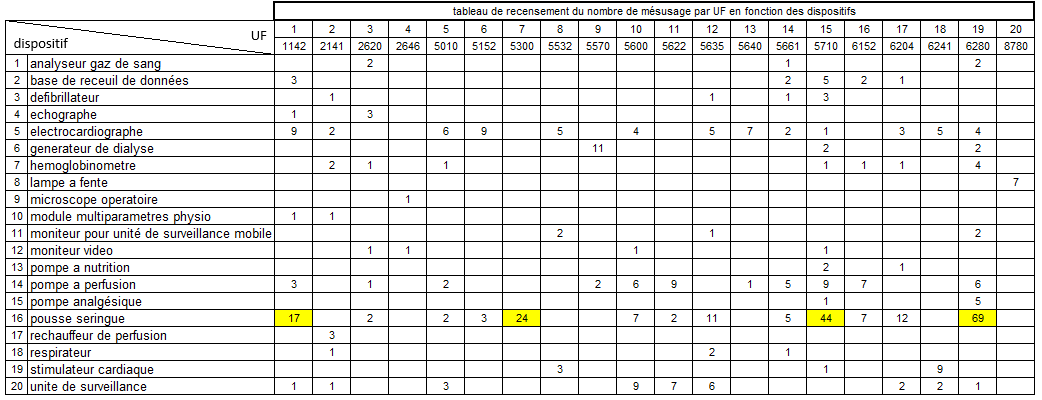
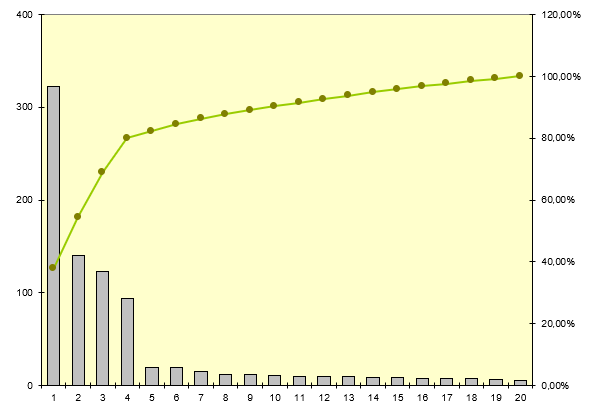
Nous allons tout d’abord définir quels sont les types de dispositif
médicaux les plus impactés par les mésusages
Ce sont des dispositifs médicaux assez commun car ils sont en
quantité relativement importantes dans les hôpitaux et employés pour
de nombres pathologies.
Certains restent néanmoins à risques potentiellement élevés pour le
patient comme les pousses seringues ou les pompes à perfusion.
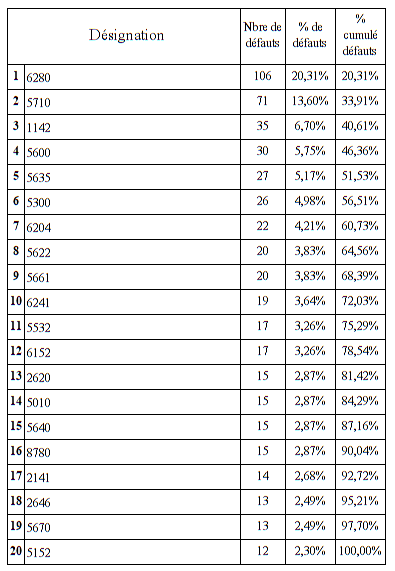
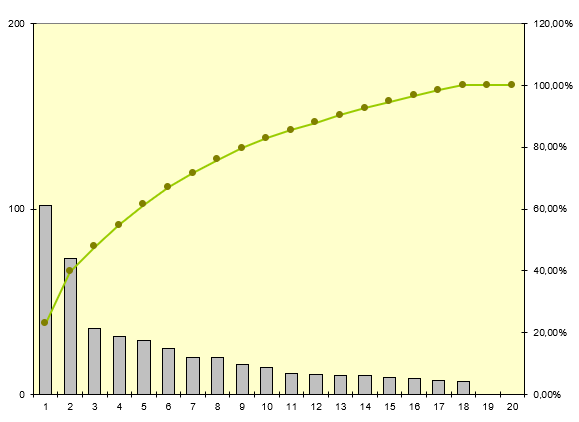
Nous allons ensuite définir les unités fonctionnelles concernées par
le plus grand nombre de mésusage
5 services concentrent plus de 50 % de l’ensemble des mésusages avec
un écart significatif entre les 2 premiers et les suivants. En
effet, ils représentent plus de 73% des mésusages des 5 premiers
services.
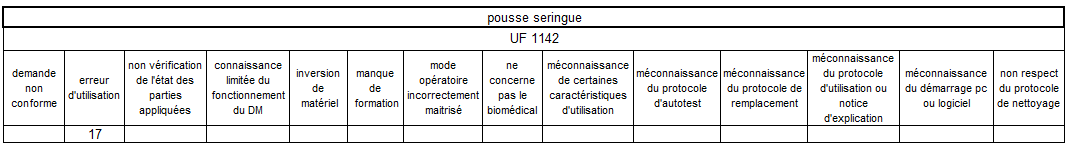
Il est à noter que l’UF
1142 (atelier biomédical) arrive en troisième position. Cela
s’explique par le fait qu’une partie non négligeable des
interventions ne sont pas déclarées par la procédure de demande
d’intervention contractuelle mais parfois par mail ou téléphone. De
ce fait, les techniciens doivent régulariser l’intervention et leur
temps de travail en déclarant eux-mêmes une intervention dans la
GMAO.
Le service biomédical rencontre également des problèmes de
déclaration de demande d’intervention effectuée par le service qui
intègre parfois plusieurs dispositifs pour une seule demande. De ce
fait, le service biomédical doit ouvrir une ou plusieurs
interventions pour régulariser cette situation.
Les deux services qui concentrent le plus de mésusage sont des
services de réanimation.
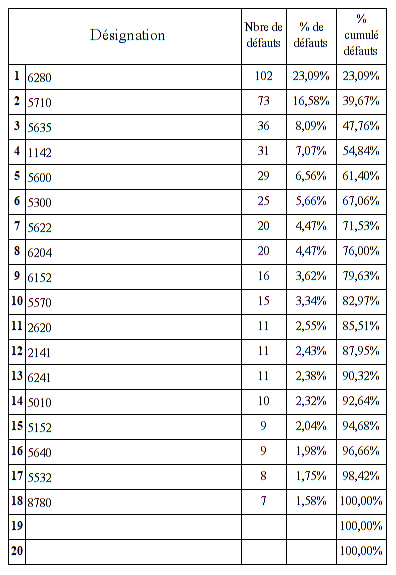
Nous allons maintenant définir la quantité de mésusage par type de
mésusage
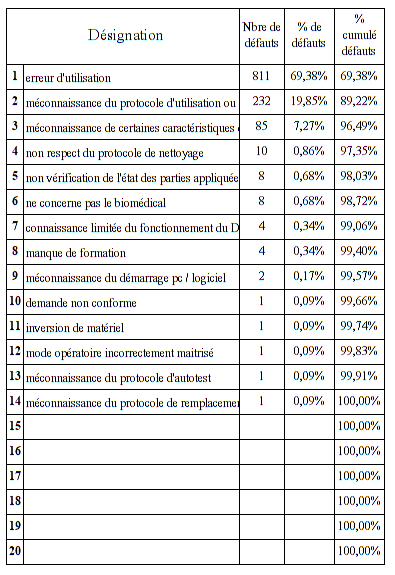
Trois mésusages représentent plus de 96 % de l’ensemble des
mésusages déclarés dans la GMAO
Erreur d’utilisation
Méconnaissance du protocole ou notice d’utilisation
Méconnaissance de certaines caractéristiques du DM
L’erreur d’utilisation reste un item généraliste qui représente 70%
des mésusages. En effet, sans précision du dysfonctionnement dans la
GMAO, il n’est pas possible d’être davantage précis sur le type de
mésusage rencontré. Ce problème survient en grande majorité quand le
technicien effectue un contrôle sur le DM et qu’il ne rencontre
aucune anomalie. (Code RAS dans la GMAO)
La méconnaissance du protocole d’utilisation et la méconnaissance de
certaines caractéristiques du dispositif sont généralement les
constats effectués par le technicien a l’issue de son diagnostic et
des échanges d’informations avec les utilisateurs. Ils représentent
environ 26% des mésusages rencontrés. Des compléments de formation
sont parfois délivrés à l’issu de ces échanges.
Nous retrouvons par ailleurs ce constat lorsque nous croisons les
données pour définir les types de mésusage en fonction des UF
recensant le plus de mésusage.
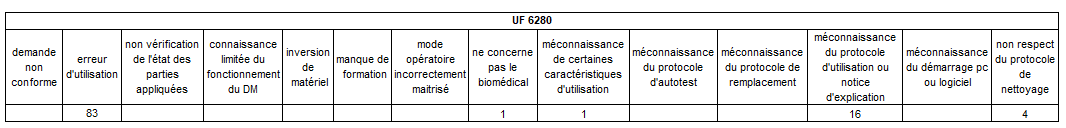
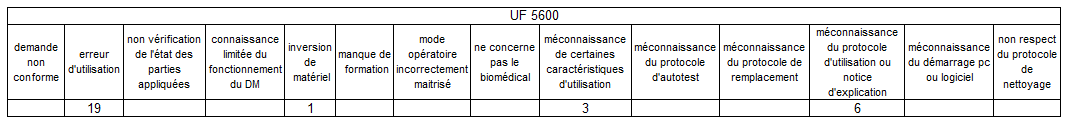
Nous allons ensuite croiser 3 requêtes pour définir sur chacun des
DM les plus impactés par le mésusage, quels sont les types de
mésusage les plus concentrés par UF.
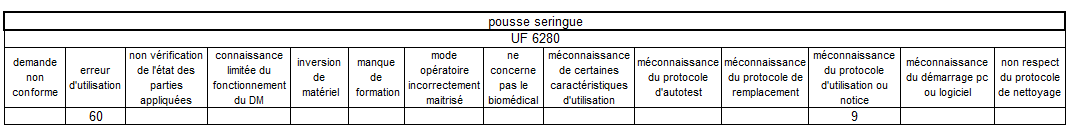
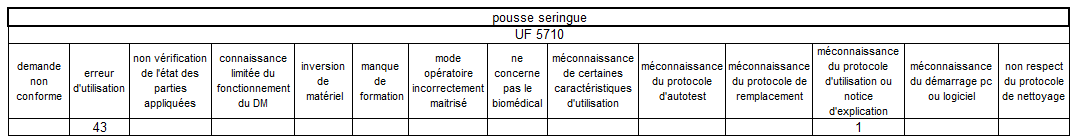
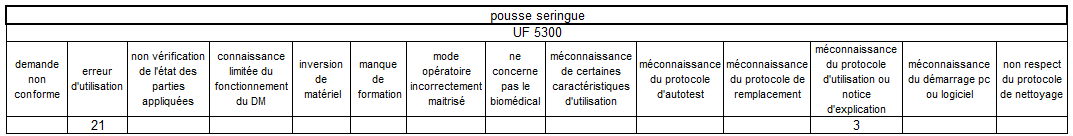
Les erreurs d’utilisation restent le mésusage largement majoritaire
sur les pousses-seringue – elles sont principalement concentrées sur
deux unités fonctionnelles de réanimation.
Les 3 autres dispositifs médicaux (pompe à perfusion, unité de
surveillance, électrocardiographe)
Il n’y a pas de lien significatif entre dispositif / UF / type de
mésusage ce qui signifie que les mésusages sont dispersés sur
l’ensemble des UF – A contrario du pousse seringue, aucun service ne
concentre un type de mésusage particulier sur un dispositif précis.
Un tableau récapitulatif nous précise de manière explicite qu’il
existe un bien un lien de cause à effet entre pousse seringue et UF
cumulant le plus de mésusage.
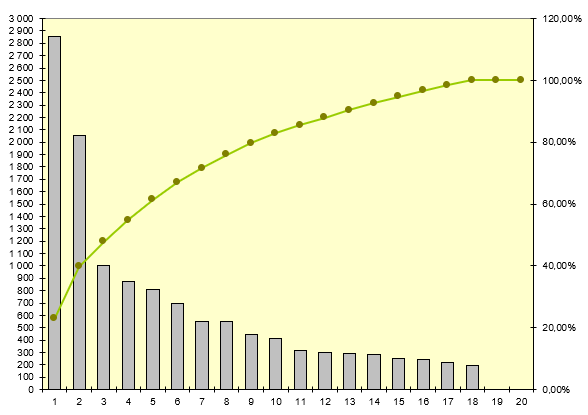
Partant de ces constatations, nous allons valoriser le temps agent
dépensé par les techniciens biomédicaux pour le traitement de ces
mésusages et le cout en temps agent induit
Nous retrouvons les 4 dispositifs les plus concernés par le mésusage
avec un temps cumulé de 322 heures pour les pousses seringue soit un
équivalent de plus de 2 mois à temps plein équivalent agent. Les
pousses seringue représentent près de 38% du temps agent dédié au
traitement des mésusages, bien loin des électrocardiographes qui
représentent environ 17% du temps agent dépensé. Il est à noter
d’ailleurs que 80% des mésusages des électrocardiographes sont dus à
des dysfonctionnements concernant l’impression au sens large du
termes (papier mis à l’envers, de travers, mauvais type de papier…)
Constat du cout en temps agent
Le cout horaire de base de calcul de 28 euros est fourni par M.
Grosjean
Le cout total en temps agent est d’environ 35 000 euros soit
l’équivalent d’un effectif temps plein avec charge. Ce cout moyen
sur 4 an représente environ 8750 euros ou un trimestre de temps
agent dédié au traitement des mésusages chaque
année
Le cout en temps agent dépensé pour le traitement des mésusages
sur les pousses seringue est valorisé à hauteur de 12 369 euros.
Quatre services concentrent à eux seuls environ 55% du temps agent
dédié aux mésusages des pousses seringue. Parmi les quatre services
concernés, l’atelier biomédical regroupe à lui seul de nombreux
services différents.
Les différentes statistiques liées aux mésusages des DM nous amènent
à plusieurs constats:
Impact du mésusage sur les DM
Le CHU de Rouen n’est pas plus impacté par le mésusage sur les DM
tel que le centre hospitalier de Pontoise par exemple qui est mon
établissement de référence. Compte tenu de l’importance de son parc
de DM, l’établissement est même sans doute légèrement moins impacté
par ce phénomène. Le management de la qualité par l’ISO 9001 et le
renforcement de la formation des utilisateurs sont sans doute des
éléments d’organisation majeurs dans la prévention des risques
néanmoins, en l’absence de Benchmark ou d’étude précise, il est
difficile d’avoir une comparaison objective d’autant que peu de
services biomédicaux hospitaliers sont certifiés , notamment sur le
même périmètre.
Ce constat ne doit pas occulter le fait que, comme d’autres
établissements, une partie non négligeable des incidents ne sont pas
déclarés par les utilisateurs.
Les dispositifs
Ainsi, les mésusages sont concentrés sur 4 dispositifs dont trois
d’entre sont des dispositifs d’usage courant (pousse-seringue, pompe
à perfusion et électrocardiographe). Parmi ces trois dispositifs,
deux d’entre-deux peuvent générer des risques cliniques
potentiellement graves pour le patient (pousse-seringue et pompe à
perfusion). Ce constat est malheureusement commun à la plupart des
établissements de santé.
Ce qui nous amène à nous poser la question de comprendre pourquoi
ces dispositifs sont constamment impactés par le mésusage de façon
générale ?
Ces dispositifs ont plusieurs caractéristiques communes à l’ensemble
des établissements de santé.
Le parc de ces dispositifs est l’un des plus conséquents parmi
l’ensemble des dispositifs des établissements de santé
Ils sont d’usage courants et fréquents
Les consommables sont diversifiés pour un même dispositif
notamment les pousses seringue
Un parc parfois hétérogène dans les établissements selon la
politique d’achat biomédicale
Le nombre de dispositifs médicaux présents au CHU de Rouende
façon conséquents tel que les pousses seringues pourrait être la
raison d'un nombre de mésusage important cependant, en appliquant
une règle de proportionnalité sur la quantité de chaque dispositif
en fonction du nombre de mésusage attribué à chaque dispositif , on
peut s’apercevoir que les dispositifs les plus concernés par le
mésusage restent ceux cités précédemment.
Les utilisateurs
Une partie importante des mésusages des pousses seringue est
concentrée sur quelques services de soins – Ce sont sans surprise
les services de réanimation dont la spécificité induit :
D’importantes équipes pouvant conduire à une diversité des
pratiques
Un travail parfois effectué dans l’urgence compte tenu des
pathologies des patients
Pour les autres dispositifs concernés, les mésusages sont dispersés
sur différents services de soins ce qui induit que le problème n’est
pas spécifique à un service ou à une pratique particulière d’autant
que ces mésusages sont communs à beaucoup d’établissements.
Le turnover du personnel de soins
Le turnover estun processus par lequel une infirmiere
quitte son poste dans un établissement ou chage de poste dans un
meme établissement. Le métier d'infirmier à l'hopital connait des
mutations importantes en raison notament du développement des
technologies, de l'informatisation, de la baisse des durée de
séjour, de la multiplication des taches administratives. Ce contexte
engendre des souhaits de mobilité plus nombreux chez le personnel de
soins , qui peuvent chercher à compenser cette intensification du
travail par la recherche d'un nouveau statut , de meilleures
conditions de travail. Les facteurs du turnover du personnel sont la
charge de travail, la rémunération, la satisfaction au travail, les
réorganisations de service et l'age. Les problémes de communication
et de stress sont également associés à un turnover élevé.
Dés lors, la transmission du savoir-faire et de l'expérience acquise
n'est plus pleinement assurée. La connaissance des dispositfs
médicaux devenus de plus en plus techniques et informatisés n'est
pas pleinement maitrisé et peu engendrer des mésusages voire des
accidents sur le patient, le personnel ou l'environnement.
Le service biomédical se retrouve alors fortement impacté par la
multiplicité des demandes d'interventions qui sont assez souvent le
fait d'une mauvaise utilisation. Les mésusages
La non connaissance de certaines caractéristiques
d’utilisation liée à un besoin de connaissance approfondie ou
spécifique du DM
Une connaissance non maitrisée du fonctionnement liée à un
manque de connaissance de base
Pour ces 2 items, plusieurs raisons peuvent être mise en évidence.
La principale est sans doute un besoin en rappel de formation
systématique ou continue.
Le troisième mésusage rencontré intitulé « erreur d’utilisation »
regroupe sans doute différents mésusages non précisément définis car
peu ou pas d’information issue de l’origine de la panne ne sont
parvenus aux techniciens biomédicaux.
Cette problématique de manque d’information dans la description du
problème ne permet pas le traitement objectif et pertinent de ce(s)
type(s) de mésusage néanmoins elle est révélatrice d’un profond
problème de communication et d’un minimum de précision de certains
utilisateurs lors de la demande d’intervention au service
biomédical.
La criticité des dispositifs médicaux
Nous avons pu déterminer que 4 dispositfs regroupaient à eux
seuls la majeure parties des mésusages néanmoins certains
dispositifs peu impactés n'en restent pas moins critiques en termes
de risques pour le patient. Il convient donc d'identifier ces
dispositfs afin de les intégrer dans les axes d'amélioration.
Trois appareils présents dans le tableau des dispositifs les plus
concernés par le mésusage sont à prendre en compte en termes de
risques.
Les générateurs de dialyse, les respirateurs et les défibrillateurs
: Dans la majeure partie des cas, ce sont des problémes de mauvaises
utilisations.
Lorsqu'il sagit d'un probléme lié à un manque de connaissance de
certaines particularités du dispositif ou lors d'une mauvaise
utilisation , les informations nécessaires à la compréhension du
probléme sont fournies par le service biomédial aux utilisateurs
sous forme de formations ou d'informations. Orientation
Ces constats nous amènent à définir plusieurs pistes de réflexion
afin de renforcer les actions existantes et de globaliser ces
actions en une stratégie unique.
En effet, une analyse uniquement focalisée sur l’exploitation des DM
en vue de minimiser les pratiques de mésusages ne suffit pas à
maitriser l’ensemble des risques qui gravitent autour du dispositif
médical.
Des actions peuvent être mises en place en amont de l’exploitation
du DM pour assurer la maitrise des risques de mésusage.
De ce fait, une analyse, théorique et pratique, des possibles pistes
d’amélioration sur l’ensemble des étapes de la vie du dispositif est
nécessaire.
Cette première étape dans la vie ou future vie du dispositif médical
est particulièrement importante en termes de gestion des risques et
maitrise des mésusages.
Nous avons effectué, dans le chapitre 1.5, la distinction entre le
mésusage sous condition normale et le mésusage sous condition
anormale d'utilisation. Le paragraphe 3.1 sera complétement dédié
aux normes nécessaires à la maitrise des mésusages des dispositifs
médicaux en utilisation normale.
Le constructeur doit garantir la conformité de son produit et de ses
activités par rapport à une liste d’exigences issues de la directive
93/42/CEE. Néanmoins, le constructeur n’a aucune obligation, selon
le produit fourni, de suivre ces exigences. Il devra néanmoins
justifier des non applicabilités.
Maîtriser les risques liés à l’utilisation du dispositif
Garantir les performances du DM
Vérifier les aspects “sécurité”
Fournir des documents d’utilisation et un étiquetage adéquat
Garantir la qualité des processus de conception, fabrication,
contrôle, transport, …
Le constructeur peut s’aider d’une série de normes pour atteindre
ces exigences. Ces normes sont donc des éléments importants à
connaitre et à vérifier dans l’optique de l’acquisition d’un
dispositif médical.
Les principales normes à disposition des fabricants
Il s’agit d’une norme reconnue comme la meilleure procédure pour
s’assurer que tous les aspects de la gestion des risques sont
évalués tout au long de la vie du dispositif médical.
En cours d’évolution, elle devrait être publiée courant 2019 - Les
principales évolutions par rapport à la version 2013 sont :
L’analyse des risques
Elle doit être assurée par le fabricant dans le cadre d’un processus
d’ingénierie d’aptitude à l’utilisation (IAU).
L’évaluation des risques
La politique d’acceptation permet de décider si le risque est à
maitriser
La maitrise des risques
La production et la conception sont listés en première intention des
options de gestion des risques
L’analyse du rapport bénéfice/risque est conditionnée par la
politique d’acceptation des risques EN 62366-1 : Ingénierie de l’aptitude à l’utilisation (IAU) [13]
L’ingénierie de l’aptitude à l’utilisation vise à maitriser les
risques dus aux erreurs d’utilisation. L’objectif est de minimiser
la probabilité d’occurrence d’une erreur d’utilisation qui peut
conduire à des risques de blessure pour les utilisateurs et/ou le
patient. Ces erreurs peuvent être causées par un problème de
perception, de cognition ou de manipulation.
Il faut pour cela que le processus de conception de l’interface
utilisateur/DM ne puisse générer d’erreur d’utilisation à chacune
de ses phases de vie notamment par la prise en compte des profils
utilisateurs, de l’environnement d’utilisation, des erreurs
d’utilisation connues et de l’amélioration connue.
Instructions d’utilisation incorrectes ou incomplètes
Alarme non perçue par l’utilisateur
Couleur d'alarme mal interprétable
Formation insuffisante de l’utilisateur
Le processus se déroule en 3 étapes
La spécification d’utilisation du DM : Il s’agit d’imaginer
tous les scénarios d’utilisation pour chaque profil utilisateur
La réalisation des évaluations formatives de l’interface : «
évaluation de l’interface utilisateur effectuée avec l’objectif
d’explorer les forces et les faiblesses de la conception de
l’interface utilisateur et les erreurs d’utilisation non prévues
». Les évaluations formatives servent à découvrir d’autres
scénarii dangereux et ainsi prendre en compte des mesures
d’amélioration de l’interface.
Effectuer une évaluation sommative : « évaluation de
l’interface utilisateur effectuée à la fin du développement de
l’interface utilisateur avec l’objectif d’obtenir une preuve
tangible que l’interface utilisateur peut être utilisée en
toute sécurité ». Il s’agit de tester tous les scénarii
dangereux dans un environnement réaliste – la documentation est
également évaluée pour vérifier si elle est compréhensible par
l’utilisateur.
EN 60601-1 : Sécurité des dispositifs électro médicaux [14]
La norme définie les exigences en matière de sécurité et de
performances essentielles applicables aux appareils électro médicaux
– Elle sert à évaluer la conformité des DM et donc à réduire les
risques.
EN ISO 13485 : Système de management de la qualité [15]
La norme permet aux fabricants de DM de satisfaire aux exigences
de la réglementation européenne. Elle permet de justifier du
marquage CE. Applicable dés qu’une assurance qualité est exigée par
la directive.
EN ISO 15223-1 : Symboles utilisés pour les instructions
d’utilisation, l’étiquetage et l’emballage [16]
L’étiquette apposée sur le dispositif doit contenir les
informations nécessaires pour que celui-ci puisse être identifié,
utilisé correctement et en toute sécurité en tenant compte de la
formation et des connaissances des utilisateurs et de permettre
d’identifier le fabricant.
EN 1041 : Informations fournies par le fabricant [17]
La norme spécifie les exigences concernant les informations à
fournir par un fabricant de dispositifs médicaux d’après la
réglementation. Cette norme est applicable au manuel utilisateur,
instruction technique, fiche…Les informations dématérialisées sont
également concernées. La norme ne spécifie pas la langue à utiliser
pour les informations ni les moyens par lesquels les informations
sont fournis.
La norme traite du développement et de la maintenance des
logiciels de dispositifs médicaux. Elle permet de mettre en place
une méthodologie délimitée et rigoureuse dont l’objectif est la
diminution des risques liés aux logiciels à un niveau acceptable. La
norme introduit 3 classes de sécurité du logiciel (A, la moins
critique à C, la plus critique).
La norme traite des bonnes pratiques cliniques applicables pour
la conception, la conduite, l’enregistrement et l’établissement des
rapports des investigation cliniques
La norme spécifie les exigences de sécurité de base et les exigences
en matière de performances ainsi que les essais des systèmes
d’alarme des appareils et des systèmes électro médicaux. Elle
définit les catégories d’alarme en termes de priorités par degrés
d’urgence, des signaux d’alarme et des états de commande cohérents
et leur marquage pour tous les systèmes d’alarme.
EN 60601-1-2 : Compatibilité électromagnétique[21]
La norme liste les exigences applicables en matière de compatibilité
électromagnétique. De plus en plus d’appareils électroniques souvent
connectés utilisent des radiofréquences qui augmentent le risque de
perturbation électromagnétique des DM. Il est donc essentiel lors de
l’acquisition d’un DM de vérifier les procédures utilisées par le
fabricant pour l’obtention de son certificat CE car elles
garantissent la sécurité d’utilisation et assurent un rapport
bénéfice/risque favorable par la mise en place de bonnes pratiques.
Une bonne connaissance des procédures utilisées par le fabricant
permettra à l’acquéreur de cibler plus précisément son besoin en
termes de gestion des risques et de lutte contre le mésusage.
La mutualisation des achats dans le secteur de la santé est
indispensable car elle est associée à la professionnalisation de
l'achat. Les spécificités des acteurs du secteur de la santé tels
que prescripteurs, utilisateurs, fournisseurs imposent une approche
réfléchie entre tous les acteurs. Chaque acteur a des problématiques
spécifiques et le modèle de chacun apporte son lot de solutions. Le
secteur biomédical n’échappe pas à cette règle. Ainsi, la sécurité,
la performance et la qualité de la prise en charge du patient
doivent être les éléments prépondérants.
L'acheteur biomédical doit donc garder sa liberté de choix en
recourant ou non à une structure de mutualisation. De plus, tout
processus de massification doit toujours permettre l'expression de
l'innovation.
Le groupement territorial d'achat (GTA) est une collaboration entre
établissements de santé pour mutualiser les besoins et procéder à
une consultation unique. Ils permettent des gains significatifs
grâce aux volumes obtenus et à l’harmonisation des besoins entre les
établissements du groupement.
La mise en place de GHT légitime la tendance de fond de
mutualisation de l’achat public et confirme les orientations prises
notamment avec l’UGAP, UNIHA ou le RESAH.
Néanmoins tout système à son lot d’avantages et d’inconvénients
Nous noterons en termes d’avantage la réduction
des coûts grâce au stockage optimisés, une optimisation des prix
de transport (moins fréquents), des réductions générales suite à
l’augmentation des volumes d’achats ou encore par une réduction
des coûts de passation de commandes. La responsabilité est
facilement identifiable en cas de situation d’achats
problématique.
Le nombre de personnes engagées dans le processus d’achats réduit le
risque d’erreur dans la passation de commande.
Une augmentation du pouvoir de négociation par effet de levier sera
effective dans la mesure où les commandes seront regroupées et
augmenteront le volume d’achat. La création d’une base commune de
fournitures et d’achats standardisés permet un meilleur suivi
comptable et une meilleure visibilité des dépenses par regroupement
des achats en un seul lieu.
A contrario, l’équipe en charge des achats centralisés peut manquer
d’expertise sur certains produits ou services et leur éloignement
par rapport aux attentes des utilisateurs peut engendrer de
l’insatisfaction.
Les avantages négociés par les acheteurs au niveau local peuvent ne
pas être à la hauteur de ceux obtenus via des achats centralisés.
Augmentation du risque de dépendance face aux fournisseurs dans la
mesure où il y a un risque d’avoir trop d’achats sur trop peu de
fournisseurs.
Stratégie d’achat
Le processus d’achat doit toujours être centré sur le besoin de
l’utilisateur. Il doit tenir compte du besoin clinique, de
l’homogénéisation des pratiques, de l’état du parc déjà présent et
des performances attendues.
Le besoin clinique est fonction de différentes données essentielles
notamment :
Le type d’unité de soins
Le type de patient
Les spécificités particulières (ambulatoire…)
Les exigences recherchées
Les consommables
Les paramétrages
….
Il est recommandé d’utiliser un même type de DM dans un même
service. Cette harmonisation contribue à la réduction des incidents
liés aux mésusages. Pour cela, un travail de concertation entre
ingénieurs biomédicaux et cadre d’unité de soins doit être un
préalable afin de gérer le parc global d’équipement de façon
optimale.
L’ingénieur biomédical détient le savoir en termes d’inventaire des
DM présent dans le service de soins. Croiser l’inventaire avec la
présence physique des DM dans le service est un préalable essentiel
avant de réfléchir aux futurs achats.
L’avis des utilisateurs en termes de besoin technique est une étape
importante dans un processus d’achat consensuel.
L’évaluation technique du dispositif médical
L’évaluation peut se faire dans le cadre du marquage CE mais
également par des essais sur banc. Les critères d’évaluation portent
sur la conception générale du DM, les modes de fonctionnements
proposés et la gamme de consommables adaptés aux besoins.
Facilité d’installation des éventuels consommables
Facilité des réglages
Lisibilité et compréhension des informations
Facilité de nettoyage et désinfection – type de produit à
utiliser
Alarmes (niveau et pertinence sonore / visuelle des catégories
d’alarme)
Les prestations et conditions de maintenance (garantie,
possibilité de prêt, mise à jour logiciel, facilité de
maintenabilité…).
La durée de la période d’essai du DM
…
D’autres critères importants sont à prendre en compte dans le choix
d’un dispositif médical ; Ils ne seront ni cités ni développés dans
ce projet car ils sont indirectement liés à la problématique du
mésusage.
La période d’essai
Une évaluation pertinente ne peut pas s’effectuer sans une période
d’essai préalable.
Pour cela, il est indispensable qu’une période d’essai du dispositif
médical en condition réelle d’utilisation dans le service
destinataire soit effectuée.
Cette période devra être suffisamment longue pour que l’ensemble des
utilisateurs puissent manipuler le dispositif et ainsi se
positionner de façon objective sur la corrélation entre besoins /
performances effectives
Le service biomédical devra s’assurer auprès du cadre de service que
des pathologies concernant la possible utilisation du DM sont
présentes au moment de la période d’essai, dans le cas d’un
dispositif particulier.
Une grille d’évaluation pourra être rédigée de façon collégiale
entre service biomédical, acteurs du service de soins et médecins.
L’avis des utilisateurs sera un atout important dans le choix final
du dispositif médical. Il permettra par ailleurs de recueillir les
premières impressions de possibilité de mésusage et validera
l’efficacité des documents mnémotechniques fournis par le
fournisseur avec le dispositif.
Il s’agit d’une étape importante qui conditionne le début de la
période de garantie du dispositif médical mais également son début
d’utilisation, d’usure et de mésusages.
Vérification des performances
Pour cela, il est essentiel de vérifier que l’appareil soit conforme
aux performances attendues. Ainsi, un certificat de contrôle qualité
en sortie de production peut être demandé au fournisseur.
Néanmoins, il est souhaitable qu’une qualification de mise en
service soit réalisée sur site afin de vérifier que le transport n’a
pas altéré les performances du dispositif. Les accessoires (cordons,
capteurs…) sont raccordés et positionnés sur le dispositif.
Le dispositif doit être paramétré avant sa mise en
service selon les recommandations du service destinataire.
Mise en place des notices et fiches mnémotechnique
La notice d’utilisation est fournie au cadre du service / personnel
utilisateur – Une fiche mnémotechnique validée lors de la période
d’essai sera placée de façon pratique et visuelle sur le dispositif
précisant les opérations basiques de condition de mise en service et
de maintenance de niveau 1. La maintenance de niveau 1 est à
effectuer par l’utilisateur.
Identification du dispositif
Lors de la mise en service, le dispositif médical est inventorié
dans le registre de sécurité, qualité et maintenance (RSQM). Une
étiquette d’identification est ainsi accolée au dispositif.
Formation des utilisateurs et techniciens biomédicaux
La formation est un point très important à évaluer dans le cadre de
l’accompagnement des soignants et techniciens biomédicaux à
l’utilisation et maintenabilité du DM.
Formation de base et rappels.
Formation spécifique des référents matériels dans le cadre de
leurs missions de gestion du parc de DM et d’interface de
communication avec le service biomédical.
Qualité des supports de formation (possibilité d’E Learning,
vidéo, manuel et notice)
Pédagogie et expérience des formateurs
Attestation de formation
Évaluation du formateur
Évaluation pratique et théorique des utilisateurs
Trois niveaux de formations sont nécessaires pour respecter la
réglementation
Une formation théorique permettant aux cadres de santé et aux
utilisateurs de connaître la réglementation et les
responsabilités pénale de chacun en cas de non-respect de son
application.
Une formation pratique initiale lors de la livraison de tout
nouveau matériel.
Une formation continue permettant aux personnels soit d’être
formés alors que le matériel est en exploitation depuis un
certain temps, soit de bénéficier d’un approfondissement sur les
modes d’utilisations des matériels après les avoir employés.
La formation continue concernant les dispositifs médicaux est
rarement organisée dans les établissements de santé. Elle est
parfois réalisée par le technicien biomédical ou dans le cadre d’un
geste commercial du fournisseur. Dans les services plus techniques
comme la réanimation médicale, ce sont les référents matériels qui
assurent parfois la formation des nouveaux arrivants.
Deux types d’évaluation pour les professionnelles doivent être
effectuées
L’évaluation de la formation portant sur la règlementation et
la politique de maintenance biomédicale
Une évaluation pour chaque utilisateur y compris les
utilisateurs arrivant après la mise en service du DM
Mise en service effective
A l’issue de ces étapes, un procès-verbal de mise en service est
rédigé et signé entre le service biomédical et le service
destinataire.
Il s’agit de la période la plus délicate car le dispositif sera
soumis à une utilisation fréquente et parfois intensive.
De nombreuses précautions afin d’éviter le mésusage ont été énoncés
dans les étapes précédent l’utilisation du dispositif cependant un
regard attentif doit être porté sur l’évolution du comportement
intrinsèque du dispositif et des pratiques d’utilisation du
personnel. Pour cela un accompagnement de la direction des soins est
impératif.
La collaboration incontournable entre direction des soins et
service biomédical.
Elle est essentielle tant pour le respect de la réglementation que
pour la mise en œuvre de la politique qualité liée au matériel
biomédical. En effet, l’appropriation des bonnes pratiques ne peut
être efficace sans une collaboration étroite, un décloisonnement du
travail et une bonne compréhension des impératifs de chacun.
Pour cela, la direction des soins doit contribuer à élaborer des
programmes de formation et détermine une politique d’évaluation des
pratiques de soins. Ces actions sont partie intégrante d’une gestion
des risques.
Ces actions doivent permettre à l’ensemble des acteurs impliqués
d’être en possession des connaissances théoriques et pratiques pour
assurer efficacement leurs missions.
Une politique qualité et gestion des risques doit être formalisée et
validée par les directions qui s’engagent en termes de moyen.
L’absence de politique qualité et le manque de concertation peuvent
entrainer des incohérences ou des interprétations d’orientation chez
les différents acteurs.
La collaboration entre les acteurs doit être formalisée notamment
sous forme de contrat de prestation. Elle engage donc les deux
parties en termes d’obligation bilatérale.
Le référent matériel
Le référent matériel est un maillon essentiel dans la lutte et la
prévention contre le mésusage. C’est une fonction qui peut être
exercée par quiconque est motivé par cette tâche. Il a un rôle
formateur lors de l’achat d’un DM, pour les nouveaux arrivants et
lors de l’évaluation des machines. Il est organisateur de la
politique de sécurité du matériel.
Cette fonction présente un aspect généraliste quels que soient les
dispositifs, portant sur la définition des procédures,
l’organisation des formations et les relations avec les
utilisateurs. Des connaissances sont spécifiques aux dispositifs ce
qui peut justifier de la spécialisation de certains référents sur
tel ou tel domaine. Le référent matériel sera sollicité lors du
choix, de l’achat et de la mise en service d’un nouveau matériel,
participer à la définition du cahier de besoins défini par le
médecin.
Il participe à l’inventaire du matériel, à la gestion du parc et
assure le suivi des accessoires, consommables des dispositifs
médicaux. Il assure la remontée d’information auprès du service
biomédical en cas de problème lors de l’utilisation.
Néanmoins le référent matériel n’est pas présent dans tous les
services et sa prestation reste tributaire de son implication et de
son efficacité. De ce fait, les formations délivrées par les
référents matériels sont très hétérogènes.
L’implication du cadre
Les cadres de santé organisent les formations des utilisateurs
surtout dans les secteurs très techniques, secteurs où les cadres
sont investis. La formation est très dépendante de la dynamique du
cadre. Il y a donc une très grande disparité de qualité de formation
entre les services.
La formation des cadres est un élément important à prendre en compte
surtout dans le domaine de la déclaration de demande d’intervention
sur un dispositif médical. La encore, la qualité et la précision de
la description du problème dans l’interface utilisateur / biomédical
de la GMAO reste dépendante de l’implication du cadre.
Les demandes d’intervention sont parfois approximatives ou
inexploitables. Des informations telles que « matériel en panne » ou
« ne fonctionne pas » sont une perte de temps pour l’ensemble des
acteurs.
Sans être réellement expert ou technicien, un peu de détails basics
sur le motif de la demande d’intervention permettrait un diagnostic
plus intuitif du technicien biomédical et un gain de temps
considérable pour l’ensemble des intervenants y compris le service
demandeur qui pourrait bénéficier d’une prise en charge et d’une
restitution parfois plus rapide de son dispositif.
Le cadre est également tributaire du retour d’information de son
personnel sur certains dysfonctionnements liés au DM. Le manque de
précision ou l’altération du message parvenu jusqu’au destinataire
constituent des demandes d’intervention peu précises et souvent
inutiles. En effet, il n’est pas rare de constater que certains DM
déposés au service biomédical sont exempts de problème. Dés lors, un
filtrage et une vérification de la pertinence des informations sont
à effectuer par le cadre avant toute demande d’intervention.
Les fiches mnémotechniques
De ce fait, il est judicieux de mettre en place sur le dispositif
médical une fiche mnémotechnique indiquant :
La maintenance de niveau 1 préalable à effectuer avant tout
appel du service biomédical
L’intervention du technicien biomédical serait ainsi conditionnée à
l’application des vérifications de base. Cette pratique limiterait
nécessairement les demandes d’intervention pour des défauts
d’utilisation trop courant tel qu’un bouton de mise sous tension
éteint ou un cordon secteur débranché.
Un livret technique pourrait également être réalisé dans chaque
service. Il aurait la particularité de recenser les DM les plus
courant d’utilisation du service et les modes opératoires de base
associés ainsi que les maintenances de niveau 1 à assurer en cas de
problème, réglages de base et protocole de nettoyage. Ce livret
pourrait être utilisé comme base pour les nouveaux arrivants mais
également en cas de perte de la fiche mnémotechnique dédiée au DM.
Ce livret pourrait être réalisé de façon collégiale entre cadre du
service de soins, direction de la qualité et service biomédical.
L’évaluation des pratiques professionnelles
Les évaluations des pratiques professionnelles relatives aux
dispositifs médicaux n’existent pas ce qui induit qu’aucun cadre
n’évalue les utilisateurs de matériel biomédical. Pourtant des EPP dans le
domaine médical ou paramédical existent notamment lors de la
certification HAS. Une EPP spécifique à l’utilisation des dispositif
médicaux et des protocoles dédiés serait sans doute un bon moyen
d’évaluer la connaissance et la maitrise des utilisateurs.
La formation continue
Il est essentiel de mettre en place une politique de formation
continue dédiée à la gestion des risques car le matériel biomédical
est souvent trop perçu comme un domaine technique réservé au service
biomédical. Or les responsabilités concernent autant les
utilisateurs que les techniciens biomédicaux en cas de défaillance.
Cette culture de la sécurité et des responsabilités doit être
présente dans l’esprit de chacun et à chaque instant.
A défaut de référent matériel, la mise en place d’un ou plusieurs
agents formés dans chaque service serait un bon relais afin
d’assurer le partage d’un savoir technique.
L’évaluation professionnelle annuelle
Un item dédié à la bonne maitrise des dispositifs médicaux pourrait
être intégré dans la grille d’évaluation annuelle de l’agent. Ainsi,
chaque agent serait orienté institutionnellement d'un besoin en
formations spécifiques sur certains dispositifs. Ce besoin devient
alors un objectif professionnel à atteindre.
Le cadre doit pouvoir prouver, au même titre que l’ingénieur
biomédical, qu’il met en œuvre les mesures réglementaires pour
s’assurer de la bonne utilisation du matériel biomédical.
Pour cela, il faut être en capacité de pouvoir mesurer que les
obligations contractualisées en fonction d’un taux « d’acceptabilité
» validé entre les deux parties ont été respectées.
Ces mesures peuvent notamment concerner le nombre de formation
demandée annuellement, la précision des demandes d’intervention
exprimée sur la GMAO, le respect du protocole de maintenance de
niveau 1. La liste n’est bien sûr pas exhaustive.
Un bilan pour chaque service pourrait être établi et supervisé lors
d’une revue de direction encadrée par le directeur des soins, le
directeur des services économiques et le directeur de la qualité.
L’interface utilisateur / biomédical
La GMAO
Une GMAO performante et dédiée aux activités biomédicales est
devenue indispensable. La traçabilité permet d’avoir une vraie
approche qualitative des événements. Elle met en évidence des pistes
d’amélioration, y compris organisationnelles. Ainsi devant une
augmentation des frais de maintenance, le service biomédical est en
mesure d’identifier les coûts services par services et d’émettre un
budget clarifié.
La capacité d’évaluer les risques de mésusage dans le temps à l’aide
des données fournies par la GMAO s’avère cruciale. Elle permet
d’ajuster par anticipation la gestion de la maintenance et la
formation pour maitriser les risques identifiés.
Une GMAO efficace contre le mésusage sera dotée d’un menu dédié au
renseignement de ces dysfonctionnements. Ce menu devra être
paramétrable par le service biomédical afin d’affiner et de filtrer
différents types de mésusage. Ce filtrage permettra de définir des
indicateurs d’alerte en cas de dépassement de seuil par exemple mais
servira également de base de travail pour les revues de direction
avec les différentes directions concernées.
L’application utilisateur de la GMAO
Elle doit être simple d’utilisation, facile et rapide d’accès,
composée de champs obligatoires à renseigner.
La mise à disposition de choix d’items par menu déroulant permet
davantage de précision lors des demandes d’intervention et permet un
jargon commun interprétable par tous.
La GMAO et son interface utilisateur permettent notamment de
formaliser et de tracer les demandes d’intervention sur un
dispositif médical. Bien que la demande d’intervention puisse se
faire de façon urgente par appel téléphonique dans des situations
particulières, elle doit néanmoins être régulariser au plus vite par
le cadre ou le référent matériel. Néanmoins, ces régularisations ne
sont pas toujours réalisées et laissent le technicien biomédical
devoir gérer cette problématique bien souvent avec peu d’information
en sa possession. Certains défauts peuvent avoir des conséquences
cliniques pour le patient.
En l’absence de déclaration, le moyen de recenser et régler ces
problématiques, est d’organiser des sessions de retour d’expérience
entre le service biomédical et les unités de soins. Cette étape
améliore la qualité et l’exhaustivité des déclarations.
Les démarches de REX (retour d’expérience) servent à tirer des
enseignements des événements indésirables associés aux pratiques de
soins. Il s’agit de présenter de manière chronologique complète,
précise et non interprétative l’événement, puis de procéder en
équipe à son analyse approfondie et systémique. Un plan d’action est
alors défini afin d’améliorer la qualité de soins et la sécurité du
patient.
Le CREX est une démarche collective d’équipe pluridisciplinaire qui
associe collecte, analyse approfondie, actions d’amélioration,
partage et communication des enseignements retirés.
C’est une démarche pour améliorer la sécurité des patients par une
analyse qui prend aussi en compte les organisations et les facteurs
humains. Elle conduit les professionnels à s’interroger en équipe
sur leurs pratiques et à prendre conscience du risque pour mieux le
maitriser.
Certains dispositifs médicaux sont soumis à obligation de
maintenance et contrôle qualité notamment les classes 2B et 3 selon
l’arrêté du 03 mars 2003. Ainsi l’exploitant est tenu d’organiser la
maintenance, qui peut être réalisée soit par le fabricant, soit par
un fournisseur de tierce maintenance, soit par l'exploitant
lui-même. A l’issue de cette maintenance préventive, le DM est
restitué au service utilisateur avec la mention de la date de la
maintenance préventive suivante explicitement portée sur le
dispositif médical par une étiquette.
Ces équipements sont soumis à des contraintes extérieures de
température, environnement électromagnétique, manipulation et
pratiques parfois inadaptées, peuvent se dérégler. Une bonne
politique de maintenance ne doit donc pas être vue comme une
contrainte réglementaire mais comme une vérification de la sûreté de
fonctionnement des équipements. La sensibilisation des services
médicaux est donc aussi importante que la bonne formation des agents
biomédicaux appliquant les protocoles de maintenance.
L’organisation de la maintenance préventive repose sur une volonté
politique de l’établissement, mais également sur des processus
définis par la politique qualité biomédicale. L’implication des
acteurs dans ces processus reste parfois encore difficile et demande
une rigueur de chacun des intervenants.
Les éléments de maintenance préventive à prendre en compte sont de
manière générale :
L’intégrité externe de l’appareil (carter, câble alimentation,
partie appliquée, support…)
Durée de fonctionnement de la batterie
Historique des alarmes
Signalisation (alarme sonore et lumineuse, afficheur…)
Le plan de maintenance préventive doit être programmé dans la GMAO
par le service biomédical dès l’acquisition de matériel selon les
préconisations du fabricant. L’organisation de la maintenance
préventive repose sur une planification du travail pour les
techniciens du service biomédical, mais également sur la mise à
disposition des matériels par les utilisateurs.[22]
Le contrôle qualité est un ensemble d’opérations destinées à évaluer
le maintien des performances définies par le fabricant. Il doit être
réalisé lors de toute maintenance préventive ou curative ou dans le
cadre
d’une acquisition de DM neuf. Un dispositif victime d’une chute doit
être vérifié par le service biomédical. Il devra subir une
maintenance et un contrôle qualité avant d’être réutilisé.
Un test de sécurité électrique est organisé afin de vérifier
l’innocuité électrique de l’appareil. L’ensemble des éléments à
contrôler sont préciser dans la norme NF EN 62 353 [23].
La maintenance corrective
La maintenance corrective rassemble les actions menées dans le but
de corriger une panne, un défaut de fonctionnement ou une chute due
à la mauvaise utilisation du DM ou un défaut inerrant à l’appareil.
Bien souvent, des demandes d’intervention pour des
dysfonctionnements relatifs à la mauvaise utilisation sont
constatées. La formation de l’utilisateur est la encore un point
essentiel car trop souvent des pannes sont créées par des défauts de
configuration. L’utilisateur génère donc lui-même une fausse panne.
Les formations, fiches mnémotechniques et procédures de maintenance
utilisateur niveau 1 mais également les stratégies d’achat comme
l’homogénéisation du parc aideront probablement à diminuer de façon
significative ce type de mésusage.
La norme NF S99-172
La norme, issue de l’ISO 9001 et de l’ISO 31000, est un système de
management du risque lié à l’exploitation des DM. Elle globalise
concept de maitrise des risques organisationnels par la maitrise de
l’ensemble des risques associés aux dispositifs médicaux.
Selon la norme NF S 99-171 [24], le service Biomédical doit
enregistrer le dispositif médical dans le RSQM.
Celui-ci permet la traçabilité des données, des opérations de
contrôle et sécurité, de contrôle qualité et de maintenance
réalisées tout au long de la vie du dispositif depuis l’achat du
dispositif médical jusqu’à sa mise en réforme ou en rebut.
Les critères de mise en réforme de DM peuvent se faire en fonction
de :
La fin d’amortissement
Le coût de maintenance cumulé par rapport au coût d’achat
initial
La notification d’obsolescence par le fabricant
L’évolution règlementaire et normative
La carence de pièces détachées et de consommables,
L’obsolescence des performances en fonction de l’évolution des
besoins
Le service utilisateur doit avoir la connaissance de la mise en
réforme de son dispositif médical de façon simple et transparente
tel que l’accès à son inventaire dans la GMAO.
Une concertation doit être effectuée entre service biomédical et
service concerné afin de préparer la planification de l’acquisition
de remplacement et de définir le budget dédié.
En règle générale, l’achat s’effectue, sauf cas particulier
d’urgence, dans le cadre du plan annuel d’investissement du service
biomédical. Le besoin doit être redéfinie et les règles dédiées à
l’étape de l’achat sont utilisées (homogénéisation du parc…).
Pendant cette période transitoire, des prêts de DM inter service
peuvent être réalisés néanmoins chaque service à la responsabilité
de vérifier les configurations de réglage nécessaires à son besoin
et de restituer dans les meilleurs délais le dispositif au service
propriétaire.
La défalcation administrative est effectuée dans le RSQM. Un
certificat de destruction est demandé au prestataire externe lorsque
le DM pris en charge n’est pas réparable – Le prestataire peut aussi
retourner le dispositif en l’état à son propriétaire. Dans ce cas,
c’est le service biomédical qui, par sa filière DEEE (déchets
d’équipements électriques et électroniques) assure la prise en
charge de la destruction du dispositif.
Étapes d’achat et de choix
de dispositifs médicaux
Premier axe
Un regard attentif doit être
apporté aux choix des normes utilisées par le constructeur du
dispositif médical en vue du marquage CE - La normeEN
62366-1 (Ingénierie de l’aptitude à l’utilisation) par exemple, permet d’augmenter la probabilité que
les utilisateurs effectueront correctement les actions sans aucune
difficulté. Le but est de réduire au maximum les risques liés à
l’utilisation normale tout en créant un outil intuitif et
efficace.
Deuxième axe
Harmoniser les pratiques d’achat et
éventuellement étendre le périmètre de certification ISO 9001 à la
fonction achat - La mise en place d'une procédure
formalisée est une étape essentielle de la professionnalisation des
achats car elle permet de maitriser les budgets. Une telle démarche
permettrait non seulement d'harmoniser les processus au sein de
l’organisation, mais surtout d’être une vitrine de diffusion des
bonnes pratiques d’achats.
Définir son besoin en coordination
avec les praticiens et utilisateurs – Une période d’essai d’une
durée suffisante pour assurer une évaluation objective du
dispositif médical par l’équipe de soins doit être privilégiée.
Quatrième axe
Prévoir des formations à
l’utilisation systématiquement et à plusieurs niveaux
Niveaux de base
« utilisateur »
Niveaux expérimenté « référent
matériel »
Niveau technique « technicien
biomédicaux »
Etape d’utilisation du
dispositif médical
Premier axe
Faire participer la direction des soins à la politique qualitébiomédicale. La direction des soins, en
partenariat avec le biomédical, doit valider les processus qualité
qui permettent aux soignants de s’approprier la réglementation et de
l’appliquer aux différentes étapes des bonnes pratiques
biomédicales.
La direction des soins doit avoir un rôle stratégique d’interface
entre le service biomédical et les services de soins. Elle doit pour
cela participer à l’élaboration des processus opérationnels telles
que :
Le processus de réception d’un
nouveau dispositif médical dans un service de soins
Le
processus de maintenance du matériel notamment l’utilisation de
la GMAO
Le
processus de contractualisation bilatérale notamment la
gestion du parc de matériel avec les modalités de mise en
service des équipements neufs, de prêt de matériel et deréforme, les
différentes maintenances et leurs modalités de mise enœuvre, la communication
et les enquêtes de satisfaction, l’organisation des formations
initiales lors de la réception de matérielneuf, lamatériovigilance.
La politique de formation - Le rôle de la direction des soins est de
permettre aux utilisateurs d’acquérir les connaissances permettant
de répondre aux exigences règlementaires sur l’utilisation des
dispositifs médicaux cependant l’initiative de la formation est
laissée au cadre de santé de chaque service de soins en fonction des
exigences et des besoins ou au service biomédical.
Un plan de formation annuel
pourrait être élaboré par le biais de la formation continue sur
les dispositifs médicaux et les services concernés mis en avant
dans les problèmes de mésusages (pousse seringue, pompe à
perfusion, centrale de surveillance, électrocardiographe) mais
également sur les dispositifs à risques pour le patient
(respirateur, générateur de dialyse et défibrillateur).
Ces formations porteraient sur larèglementation, l’utilisation du matériel lors de sa 1ère
mise enservice, un
diagnostic de niveau 1 préalable avant toute demande d’intervention
au biomédical en cas de problème, une mise en pratique lors des
recrutements de nouveauxpersonnels
et un approfondissement dans l’utilisation de matérielcomplexe.
Troisième axe
La mise en place de référent
matériel dans les secteurs particulièrement concernés par le
mésusage – Après quelquesmois d’utilisation et
d’appropriation du matériel par les professionnels, de nouvelles
questions se posent, des automatismes s'installent, des
connaissances se perdent, le personnel arrivant aprés la mise en
service du matériel n'est pas formé, le turnover des équipes est
parfois important...la mise en place de référent matériel formateur
permettraient d'assurer la transmission du savoir-faire et
d'homogénéiser les pratiques. Il est néanmoins indispensable que ces
référents puissent bénéficier de formations spécifiques renforcées
et cela chaque année.
Quatrième axe L’évaluation des pratiques
professionnelles spécifique à l’utilisation des dispositifs
médicaux – Les dispositifs médicaux sont au cœur de la
problématique de la sécurité et de la qualité des soins.
L’évaluation de la maîtrise des risques pour les utilisateurs est
donc devenue une exigence essentielle. La formalisation d’une
politique d’évaluation des pratiques paramédicales permettrait de
répondre à cette exigence.
Etape de
maintenances et contrôles
Les maintenances préventives et
curatives - L’organisation repose sur des processus
définis par la politique qualité biomédicale. L’implication des
acteurs dans ces processus demande une rigueur et une
responsabilisation de chacun. L’organisationnelle repose sur la GMAO
et sur une organisation mensuelle des maintenancespréventives. La
collaborationentreleservicebiomédicaletlesservicesdesoins doit être efficace.
Elle repose notamment sur la précision de la formulation des
demandes d’intervention dans la GMAO, souvent trop imprécises, le
respect des clauses du contrat bilatérale et sur la qualité et
l’efficacité des réponses apportées par le service biomédical aux
utilisateurs.
Le mésusage a un réel impact en termes de difficultés de maitrise de
gestion des risques et de couts induits.
Il nécessite un effort de lutte permanent et doit s’inscrire de
manière continue dans la mise en place de méthodes et de moyens à
chaque étape de la vie du dispositif médical.
En effet, certaines actions peuvent être limitées en efficacité dès
lors que le périmètre d’intervention se situe uniquement au niveau
de l’exploitation.
La gestion des risques s’effectue stratégiquement, de façon
globalisée dans le processus de vie du DM, mais des actions ciblées
et spécifiques doivent être mise en place parfois de façon
renforcées sur certains DM ou les problèmes de mésusages sont
récurrents.
Le CHU de Rouen n’échappe pas à cette problématique bien que le
nombre de mésusage recensé en fonction du nombre de dispositif en
exploitation soit relativement maitrisé. Les actions menées par le
service biomédical depuis plusieurs années dans le cadre de la
certification ISO 9001 sont sans doute les éléments frein à une
inflation des mésusages.
Dans un contexte où les mésusages sont focalisés sur des problèmes
d’utilisation, la direction des soins devient un acteur
incontournable pour impulser une dynamique auprès des cadres de
santé et du personnel de soins.
Le directeur des soins est donc un acteur important de la définition
de la politique qualité et gestion des risques liées à l’utilisation
du matériel. Il doit s’impliquer dans la mobilisation de tous les
acteurs de soins et dans leur formation, afin de ne pas exposer le
personnel à d’éventuelles poursuites judiciaires en cas d’incidents
cliniques liés aux mésusages.
Annexe
1 : Carte du groupement CHU de Rouen - source site CHU Rouen Annexe
2 : Organigramme fonctionnel du CHU de Rouen - Source
service biomédical Rouen Annexe
3 : Équipe des techniciens biomédicaux du CHU de Rouen -
source service biomédical Rouen Annexe
4 : Tableau d’inventaire des DM du CHU de Rouen - source
service biomédical Rouen Annexe
5 : Tableau du nombre d’intervention par type de DM - source
service biomédical Rouen Annexe
6 : Cartographie des processus - source service biomédical
Rouen Annexe
7 : Fiche de déclaration de matériovigilance - source site
ANSM Annexe
8 : Questionnaire d’audit pour les ingénieurs - réalisation
personnelle Annexe
9 : Questions / réponses Questionnaire d’audit - réalisation
personnelle Annexe
10 : Divers travaux participatifs de maintenances
préventives et curatives sur les respirateurs - réalisation
personnelle Annexe 11 :
Réalisation d'un contrôle qualité échographe + rédaction des modes
opératoires de test de résistance d'isolement et courant de fuite
sur sondes - réalisation personnelle
Remarques :
Les
réponses
aux questions de l’audit sont le reflet de la tendance
générale des personnes auditées
- Les réponses des personnes auditées sont en bleues italiques afin de faciliter
la lecture
Questionnaire dédié au service de soins - Catégorie de personnel
auditée : référent matériel
1-Bénéficiez-vous d’une période d’essai des DM avant le choix final
du matériel ?
Oui pour chaque nouveau dispositif – les
médecins évaluent d’abord le matériel puis le cadre et ensuite
les utilisateurs (infirmière, aide-soignant). La période dure 1
semaine par DM néanmoins celle-ci est parfois trop courte car
l’ensemble du personnel n’est pas toujours présent (astreintes,
congés) et les pathologies présentes au moment des essais ne
correspondent pas toujours à la finalité du DM – une grille
d’évaluation est remplie par plusieurs soignants afin de donner
un avis collégial sur la facilité d’utilisation.
2-Suivez-vous systématiquement une formation du fournisseur lors de
l’acquisition d’un dm ?
Oui, la formation est toujours assurée
3-Les formations vous semblent-elles claires et efficaces ?
Oui – De plus, les fournisseurs assurent la
formation sur toutes les périodes de présence de chaque
personnel
4-Prenez-vous le temps de lire les notices d’utilisation des DM que
vous utilisez ?
Pas toujours – cela dépend des urgences à
traiter
5-Les notices d’utilisation des DM vous semble t’elles faciles
à lire ?
Oui pour certaines mais parfois une fiche mnémotechnique qui
résume les fonctionnalités de base serait beaucoup plus pratique
6-Les nouveaux arrivants sont-ils formés au fonctionnement des DM ?
Par qui ?
Ils bénéficient d’une formation de 5 à 6
semaines avec un tuteur (infirmier ou cadre) – ils peuvent
bénéficier de 2 semaines supplémentaires à leur demande si
besoin.
7-Une évaluation sur la bonne utilisation des DM est-elle pratiquée
?
Oui, à la fin de la période tutorale, une
évaluation est effectuée par le cadre de service.
8-Les dysfonctionnements liés aux DM sont-ils systématiquement
déclarés ? Oui sauf si nous les réglons de
nous-mêmes
9-Faite vous la distinction entre une FEI et une déclaration MV ?
Oui. En cas de doute, nous avons
l’application GEDI
pour nous aider
10-Selon vous, de façon générale, qu’elles aspects pourraient être
améliorés pour faciliter l’utilisation des DM ?
Principalement l’ergonomie de certains
dispositifs, le poids et l’intuitivité
11-La communication avec le service biomédicale vous semble-t-elle
efficace ?
12-Un référent matériel vous semblerait-il une interface essentielle
entre le service biomédical et le service de soins. Pourquoi ?
Oui, car nous apportons les informations
complémentaires nécessaires aux soignants comme au service
biomédical mais nous avons repris petit à petit une partie du
travail que le cadre ne pouvait plus assurer ce qui nous laisse
moins de temps pour gérer ces missions.
Questionnaire dédié aux techniciens biomédicaux
1- Quels sont les DM dans votre domaine de compétence les plus
concernés par les problèmes d’utilisation ?
Électrocardiographe et la surveillance
cardiaque – pousse seringue et pompe à perfusion - Bistouri
électrique et colonne de cœlioscopie Certains techniciens ne sont pas ou peu impactés dans leur
domaine d’intervention par le mésusage
2-Quels sont les types de mauvaise utilisation les plus courants ?
Ils sont liés aux utilisations de
consommables ou parties appliquées, à la mauvaise connaissance
du DM ou de certaines de ses fonctionnalités et également aux
inattentions.
3-A quelle fréquence rencontrez-vous ces mésusages ?
Les fréquences sont variables – elles
dépendent en fait de l’utilisateur
4-Quels sont les impacts sur votre travail au quotidien ?
Ces mésusages ont surtout un impact sur
notre temps de travail (intervention + traçabilité +
communication) mais également sur notre moral – nous avons
parfois l’impression de répéter les mêmes explications pour les
mêmes problèmes.
5-Quels sont les moyens mis en œuvre à votre niveau pour éviter la
mauvaise utilisation ?
La formation et la sensibilisation sont les
seules réponses dans l’immédiat à notre niveau de technicien –
les formations sont parfois vite oubliées
6-Les moyens mis en place vous semblent-ils efficaces ?
Pas toujours car il y a une certaine
récurrence sur la mauvaise utilisation de certains DM - Des
évaluations sur les formations sont effectuées mais les
problèmes sont plutôt personnel-dépendant – les professionnels
de soins n’ont pas toujours conscience du danger lié aux
mésusages des DM
6-Pensez-vous que d’autres moyens pourraient être mis en œuvre pour
éviter les mésusages ?
Certains cadres devraient être davantage
impliqués – une proposition de formation lui est faite et c’est
lui qui accepte ou pas – ce n’est pas toujours accepté et cela
reste parfois sans réponse – Des documents mnémotechniques sur
les DM concernés expliquant la maintenance de niveau 1 et les
fonctionnalités de base nous éviterait sans doute des
interventions inutiles.
7-Pensez-vous qu’un item spécifique aux mésusages devrait apparaitre
dans la prochaine GMAO ?
Oui car cela nous permettrait notamment
d’affiner les problèmes liés aux mésusages
8-Pensez-vous qu’un référent matériel soit un moyen efficace de
lutter contre le mésusage ? pourquoi ?
Oui, c’est un gain de temps appréciable –
ce sont généralement des aides-soignantes / cadre tech de labo
mais la prestation reste personnel dépendant. Le problème est
qu’elles ne sont pas exclusivement dédiées à part entière à
cette fonction car elles assurent également la logistique, les
inventaires et les états des stock des consommables entre
autres. Cette fonction reste également personnel-dépendant. Le
réfèrent matériel assure la communication avec le service
biomédical et les soignants car les équipes ne se transmettent
pas toujours les informations.
Questionnaire dédié aux ingénieurs biomédicaux
1-Quelle est votre politique d’achat des DM ?
Principalement par centrale d’achat
(directive DGOS) sinon procédure d’achat type MAPA ou AO
2-Quels sont les principaux critères de choix de vos dispositifs
médicaux ?
Ils sont axés sur le besoin de
l’utilisateur avant tout mais également sur l’homogénéité du
parc, la gestion des risques et le cout entre autres.
3-Le mésusage au sens large tient-il une place importante dans votre
choix ?
Non, ce n’est pas un critère essentiel
néanmoins nous essayons de le minimiser grâce à la mise en place
de période d’essai de matériel, de formation et de gestion des
risques.
4-Une période d’essai des dispositifs médicaux dans les unités est
donc organisée avant votre choix ? Combien de temps dure-t-elle ?
Elle dure environ 1 semaine la plupart du
temps
5-Une évaluation des utilisateurs est-elle effectuée après la
période d’essai ? Oui – cette évaluation est importante car elle nous sert à
affiner notre choix
6-Des formations du fournisseur sur l’utilisation du matériel
sont-elles systématiquement demandées dans vos exigences ?
Non pas systématiquement – si le matériel
est déjà existant, nous ne demandons pas de formation – si le
matériel est nouveau alors c’est une exigence – Néanmoins le
cadre peut nous faire des demandes de formation s’il en estime
le besoin.
7-Selon vous, quelle devrait être la fréquence nécessaire de mise à
niveau en formation pour le personnel utilisateur ?
Difficile de répondre ; Cela dépend des
types de matériels et des services – les fréquences seront
nécessairement différentes en fonction de ces caractéristiques.
8-Avez-vous un retour mesurable sur la pertinence de formation des
utilisateurs par les techniciens biomédicaux ou cadre de service ?
Un bilan de satisfaction des prestations
auprès des cadres de pole est effectué tous les 2 ans.
9-Pensez-vous intégrer un item spécifique aux mésusages dans votre
nouvelle GMAO ?
Il me semble qu’il existe un item
spécifique dans un menu déroulant mais il est généraliste
10-Faite vous remonter aux fournisseurs les mésusages rencontrées
sur leur DM ?
Oui s’ils sont récurrents
11-Faite vous un benchmark avant le choix de vos DM afin de
connaitre les forces et faiblesses de vos futurs DM en termes de
mésusages a priori ?
Oui de temps en temps sinon nous demandons
plutôt à nos collègues ingénieurs
Questionnaire dédié aux fournisseurs/prestataires biomédicaux
1-Quelle sont les types de mésusage auxquels vous êtes confrontés ? Ce sont principalement des défauts d’utilisation liés au
nettoyage ou à la désinfection des dispositifs
2-Avez-vous des exemples précis ?
Lors du nettoyage des parties appliquées,
les cordons ne sont pas débranchés du dispositif. Ils sont
nettoyés puis enroulés tels quels ce qui entraine un phénomène
de torsion du câble - les fils finissent par s’arracher ou les
connectiques finissent par se dessouder Certains blocs patients de respirateur se stérilisent en
autoclave mais ils doivent être préalablement démontés car
certaines pièces ne sont pas autoclavables. Il arrive que le
bloc patient soit directement autoclavé sans démontage ce qui
entraine une dilatation des matériaux et génère des fuites sur
le respirateur – ces pannes sont très difficiles à diagnostiquer
3-Que préconise le constructeur pour éviter ces mésusages ?
Des recommandations sont stipulées dans la
notice d’utilisation. Des produits de nettoyage/désinfection
sont préconisés/recommandés par le fournisseur néanmoins ces
produits sont généralement issus du pays du constructeur – il
est donc impératif d’utilisé le produit équivalent molécule dans
le pays utilisateur – Les hygiénistes des hôpitaux recommandent
d’utiliser le produit en marché dans l’établissement et cela
n’est parfois pas compatible avec l’intégrité à plus ou moins
long terme du DM.
4-Délivrez-vous une formation aux utilisateurs lors de la fourniture
d’un dispositif médical ?
Oui, c’est le travail de l’ingénieur
d’application – il effectue au préalable une démonstration
devant les utilisateurs et le service biomédical – il revient
ensuite pour assurer une formation en fonction du besoin de
chacun
Une configuration des dispositifs (exemple des seuils d’alarme)
est effectuée avec le cadre et le médecin car les demandent sont
spécifiques à chaque discipline.
5-Pensez-vous qu’une seule formation soit efficace en termes
d’utilisation de DM ?
Non, on s’aperçoit que le besoin de mise à
niveau est souvent essentiel dans la pérennité du matériel –
Nous fournissons une formation de base ; après le service
biomédical prend le relais
6-Pensez-vous que le service biomédical soit compétent en tant que
formateur ?
Oui, ils connaissent et maitrisent les
dispositifs dont ils ont la charge mais ce ne sont pas des
formateurs à proprement parlé – ils sont techniciens avant tout.
La formation est un véritable métier qui demande notamment de la
pédagogie et la capacité de se mettre au niveau de l’ensemble
des utilisateurs.
7-Quelle est selon vous les causes potentiellement récurrentes de
mésusage malgré la délivrance de formation ?
Les hôpitaux sont confrontés à beaucoup de
turn over du personnel de soins – parfois la transmission du
savoir-faire n’est pas effectuée et les nouveaux arrivants ne
savent pas se servir du matériel – j’ai déjà été confronté à
cette situation lors d’un achat complémentaire de télémétrie.
Les matériels n’ont jamais été utilisés car le personnel formé
initialement sur cette nouvelle technologie n’était plus dans
l’établissement et n’avait pas formé les nouveaux arrivants.
8-Comment est gérée la matériovigilance dans votre société ?
Le client doit nous faire une demande
d’intervention obligatoirement car sans cela nous n’intervenons
pas. En effet, même s’il s’agit d’une déclaration de
matériovigilance, l’intervention est facturable. Dans 95% des
déclarations, le problème mentionné est d’ordre d’un problème
d’utilisation. Nous faisons une enquête et nous assurons une
extraction des logs du dispositif. Le dispositif est ensuite
envoyé pour expertise auprès de l’ingénieur chargé de traiter
les dossiers matériovigilance
9-Quel type de matériovigilance rencontrez-vous le plus souvent ?
En fait, il s’agit de mésusage la plupart
du temps – souvent il s’agit de problème d’alarme qui soi-disant
ne fonctionnent pas – dans la plupart des cas, les alarmes ont
été acquittées par le personnel en position silence (alarme
totale).
Annexe 10 : Divers travaux participatifs
de maintenances préventives et curatives sur les respirateurs
Maintenance curative sur respirateur d’urgence Monnal T75 Air
liquide
Travaux effectués : Mise en place d’un rehausseur sur bloc valve
d’insufflation – calibrage du dispositif – remplacement de la
batterie – mise a jour de la version du logiciel – autotest
Démontage des carters du ventilateur - Repérage
et déconnexion des flexibles pneumatiques sur la carte mère
Dépose du bloc d’alimentation et mise en place
du rehausseur sur le bloc valve insufflation - remise en place du
bloc alimentation + chambre de mélange des gaz
Connexion électrique et pneumatique de la carte
mère
Mise sous tension du ventilateur en mode VS/AI
(ventilation spontanée avec aide inspiratoire et PEP)
Une pression positive constante (AI) est maintenue au-dessus du
niveau de la PEP dans le circuit patient à chaque effort
inspiratoire du patient. Le passage en phase expiratoire peut être
déclenché
- Si le débit devient inférieur au seuil
expiratoire fixé
- Par un effort expiratoire du patient
- Par atteinte de la durée maximale d’insufflation
réglée
Étalonnage des capteurs de débit et pression en mode maintenance
Utilisation du Système de contrôle de
ventilateurs « Certifier FA Plus 4080 » pour l’analyse de débit de
gaz.
Cet ECME (équipement de contrôle, mesure et essai) contrôle
quasiment tous les modèles de ventilateurs : adulte, pédiatrie,
anesthésie, néonatologie et haute fréquence. Il peut également être
utilisé pour tester divers équipements médicaux, notamment des
doseurs de gaz anesthésiants, des insufflateurs et des
concentrateurs d'oxygène.
Confirmation d’étalonnage
réussi
Mise à jour de la version software du ventilateur avec application «
Monnal software management ». Message de configuration réussi
Remplacement de la batterie et auto test du dispositif – Mise en
fonctionnement du respirateur pendant quelques heures avec
réglages paramètres + mise en charge de la batterie
Problématique signalée par le service : alarme de « défaut débit »
lors de la mise en place du capteur.
Après essais, le connecteur du capteur de débit semble être la cause
du déclenchement intempestif de l’alarme du ventilateur.
Démontage du ventilateur - séparation de la
partie électronique et pneumatique.
Remontage du ventilateur et démarrage de
l’autotest. Mise en fonctionnement en mode VAC avec réglage FIO2,
FC, P crête, P plateau, PEP pendant quelques heures pour
vérification.
Dépannage du respirateur de réanimation Servo i
Problématique signalée par le service : alarme de « transducteur
pression » lors de l’autotest du ventilateur
Dépose du capteur de pression et contrôle de
l’état de la membrane - Constat : membrane percée =>
remplacement de la membrane
Réglage des paramètres en mode volume contrôlé
– FIO2, PEP, fréquence cardiaque et volume courant - Mise en
fonctionnement pendant quelques heures avant restitution
Maintenance préventive sur respirateur anesthésie Primus -
Remplacement du kit 6 ans
Démontage du bloc patient - Remplacement des
filtres et joints sur arrivées des gaz (protoxyde, oxygène, air) –
remplacement du nafion et des joints toriques
Travail inspiré du rapport UTC "Contrôle Qualité en échographie avec
élaboration d'un Travail Pratique" , Dominique FRANCOISE, François
NILINGIYIMANA, Patrice RICHAUD, Christophe VILA, Projet,
Certification Professionnelle ABIH, UTC, 2011